Data set from Luis Lab
Dongyue Xie
2021-08-25
Last updated: 2021-09-10
Checks: 7 0
Knit directory: SMF/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190719) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version ed30472. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/external_data/
Untracked files:
Untracked: analysis/sparse_smoothing.Rmd
Untracked: code/wave_ebmf.R
Untracked: code/wave_ebmf_run.R
Untracked: data/luis/
Untracked: output/flash_on_cmc2_reduced.rds
Untracked: output/flash_on_cmc2_reduced_2000kb.rds
Untracked: output/wave_flash_on_cmc2_reduced_2000kb.rds
Unstaged changes:
Modified: code/smooth_flash.R
Modified: code/sparseWS.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/luis_data.Rmd) and HTML (docs/luis_data.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | ed30472 | Dongyue Xie | 2021-09-10 | Build site. |
| Rmd | 52cea82 | Dongyue Xie | 2021-09-10 | wflow_publish(“analysis/luis_data.Rmd”) |
| html | 826e06f | Dongyue Xie | 2021-09-10 | Build site. |
| Rmd | 374e2fb | Dongyue Xie | 2021-09-10 | wflow_publish(“analysis/luis_data.Rmd”) |
| html | 5450d94 | Dongyue Xie | 2021-09-10 | Build site. |
| Rmd | c187576 | Dongyue Xie | 2021-09-10 | wflow_publish(“analysis/luis_data.Rmd”) |
Introduction
“The idea is that gene expression at a given gene might be associated wit the marks in parts of the genome near-by that gene. But we don’t know in advance exactly where to look for that signal - just nearby the gene. And the signal might be at different places for different marks or the same place.”
“So by running factor analysis I hope to automatically identify factors representing the things that covary… eg co-variation of gene expression with certain marks”
Try to look at some data examples.
CMC2
CMC2 is a protein coding gene, with 10 exons.
library(readr)
CMC2_ATACseq = read_csv("data/luis/CMC2_ATACseq.csv")Warning: Missing column names filled in: 'X1' [1]
── Column specification ────────────────────────────────────────────────────────
cols(
.default = col_double(),
locus = col_character()
)
ℹ Use `spec()` for the full column specifications.CMC2_RNAseq = read_csv("data/luis/CMC2_RNAseq.csv")Warning: Missing column names filled in: 'X1' [1]
── Column specification ────────────────────────────────────────────────────────
cols(
.default = col_double(),
locus = col_character()
)
ℹ Use `spec()` for the full column specifications.CMC2_H3K4me1 = read_csv("data/luis/CMC2_H3K4me1.csv")Warning: Missing column names filled in: 'X1' [1]
── Column specification ────────────────────────────────────────────────────────
cols(
.default = col_double(),
locus = col_character()
)
ℹ Use `spec()` for the full column specifications.CMC2_ATACseq = do.call(cbind.data.frame, CMC2_ATACseq)
CMC2_RNAseq = do.call(cbind.data.frame, CMC2_RNAseq)
CMC2_H3K4me1 = do.call(cbind.data.frame, CMC2_H3K4me1)
dim(CMC2_ATACseq)[1] 426090 72dim(CMC2_RNAseq)[1] 426090 80dim(CMC2_H3K4me1)[1] 426090 56What do data look like?
plot(CMC2_ATACseq$AF08_Flu,type='l',xlab = 'locus',ylab='',main = 'CMC2 ATAC AF08_Flu')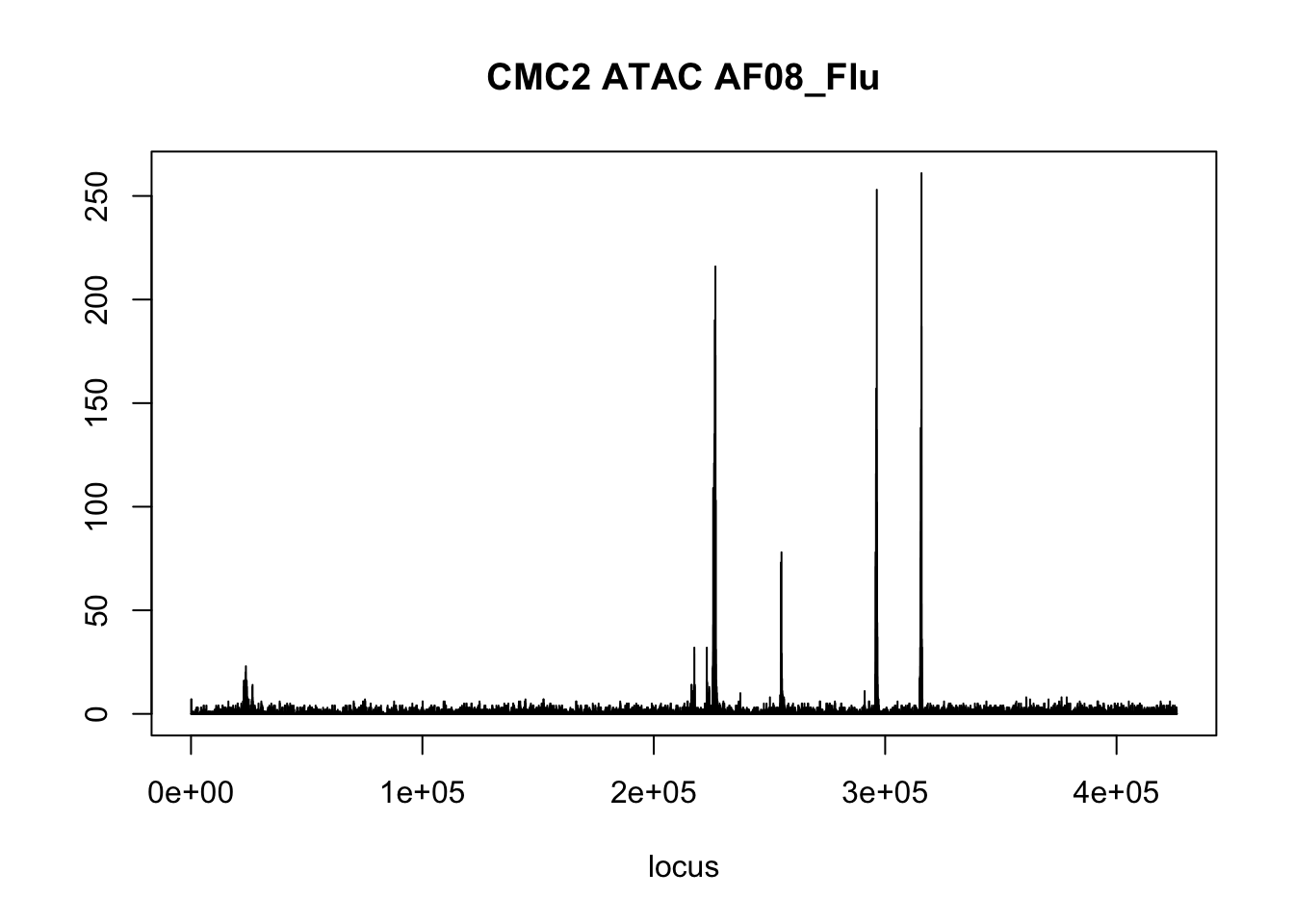
| Version | Author | Date |
|---|---|---|
| 5450d94 | Dongyue Xie | 2021-09-10 |
plot(CMC2_RNAseq$AF08_Flu,type='l',xlab = 'locus',ylab='',main = 'CMC2 RNA AF08_Flu')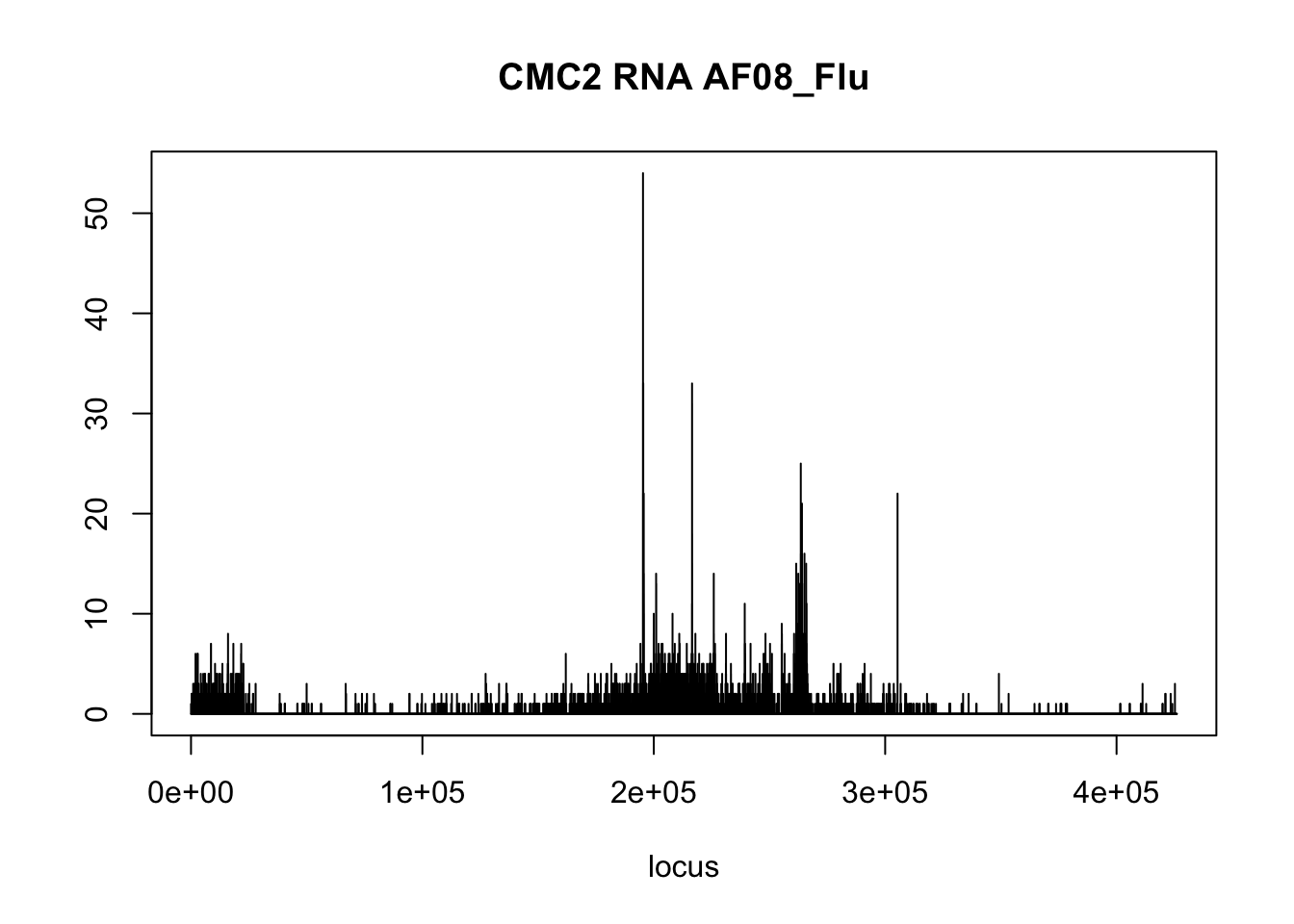
| Version | Author | Date |
|---|---|---|
| 5450d94 | Dongyue Xie | 2021-09-10 |
plot(CMC2_H3K4me1$AF08_Flu_H3K4me1,type='l',xlab = 'locus',ylab='',main = 'CMC2 H3K4me1 AF08_Flu')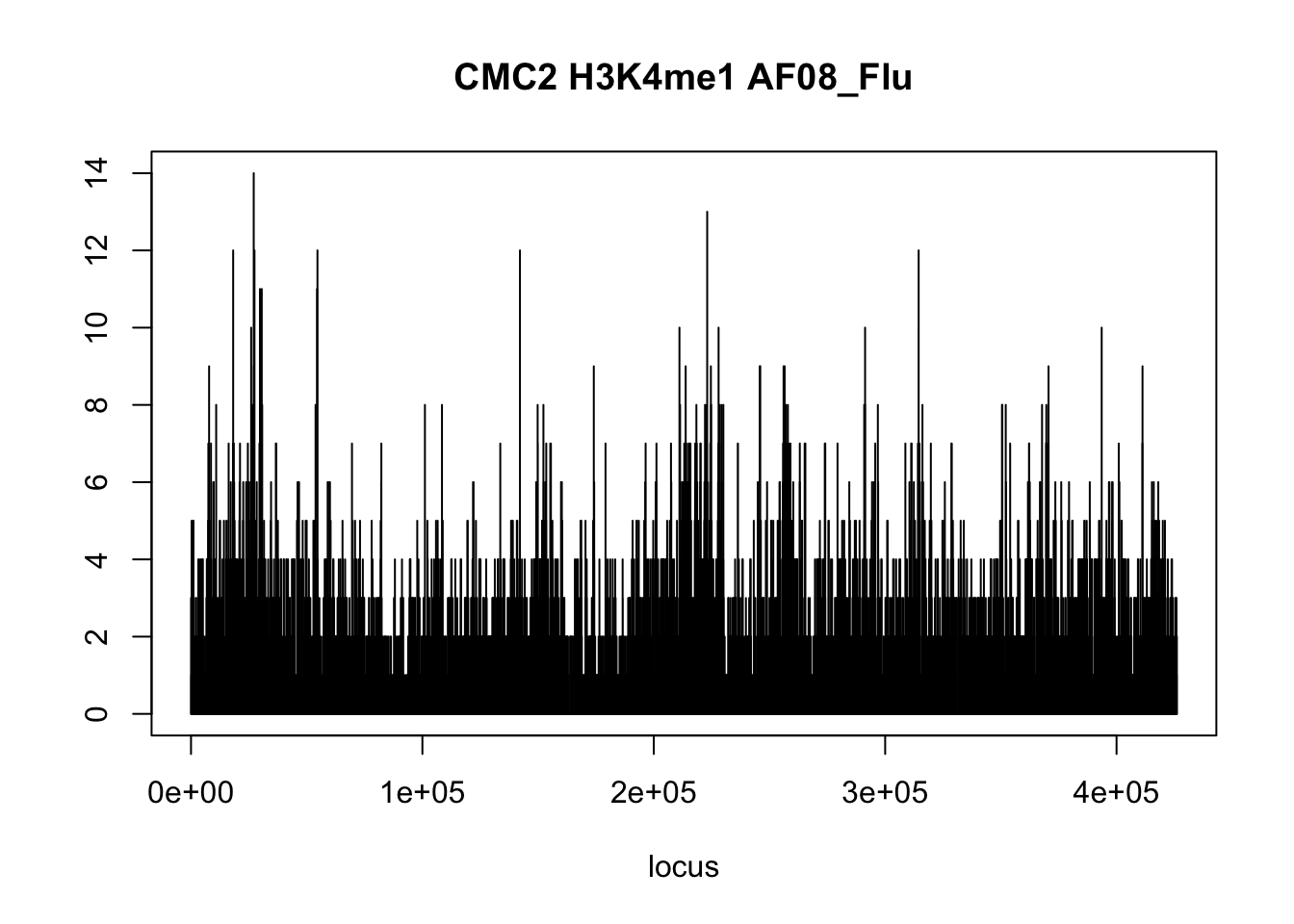
| Version | Author | Date |
|---|---|---|
| 5450d94 | Dongyue Xie | 2021-09-10 |
How sparse the data are?
sparse_prop = function(Y){
sum(Y==0)/prod(dim(Y))
}
sparse_prop(CMC2_ATACseq[,-c(1,2)])[1] 0.9674908sparse_prop(CMC2_RNAseq[,-c(1,2)])[1] 0.9738769sparse_prop(CMC2_H3K4me1[,-c(1,2)])[1] 0.9594024Get overlapping individuals
indis_ATAC = (colnames(CMC2_ATACseq))[-c(1,2)]
indis_RNA = (colnames(CMC2_RNAseq))[-c(1,2)]
indis_H3K4me1 = (colnames(CMC2_H3K4me1))[-c(1,2)]
for(i in 1:length(indis_H3K4me1)){
name_i = strsplit(indis_H3K4me1[i],split = '_')[[1]]
indis_H3K4me1[i] = paste(name_i[1],name_i[2],sep = '_')
}
indis = intersect(intersect(indis_ATAC,indis_RNA),indis_H3K4me1)
indis [1] "AF08_Flu" "AF08_NI" "AF12_Flu" "AF12_NI" "AF16_Flu" "AF16_NI"
[7] "AF18_Flu" "AF18_NI" "AF20_Flu" "AF20_NI" "AF22_Flu" "AF22_NI"
[13] "AF24_Flu" "AF24_NI" "AF26_Flu" "AF26_NI" "AF28_Flu" "AF28_NI"
[19] "AF30_Flu" "AF30_NI" "AF34_Flu" "AF34_NI" "AF36_Flu" "AF36_NI"
[25] "AF38_Flu" "AF38_NI" "EU03_Flu" "EU03_NI" "EU05_Flu" "EU05_NI"
[31] "EU07_Flu" "EU07_NI" "EU09_Flu" "EU09_NI" "EU13_Flu" "EU13_NI"
[37] "EU15_Flu" "EU15_NI" "EU19_Flu" "EU19_NI" "EU21_Flu" "EU21_NI"
[43] "EU25_Flu" "EU25_NI" "EU27_Flu" "EU27_NI" "EU33_Flu" "EU33_NI"
[49] "EU39_Flu" "EU39_NI" "EU41_Flu" "EU41_NI" "EU47_Flu" "EU47_NI" length(indis)[1] 54Only use NI individuals.
indis_NI = c()
for(i in 1:length(indis)){
id = strsplit(indis[i],split='_')[[1]][2]
if(id=='NI'){
indis_NI = c(indis_NI,indis[i])
}
}Try to run flashier on [RNA seq, H3K4me1, ATAC]
Y_RNA = t(CMC2_RNAseq[,match(indis_NI,indis_RNA)+2])
Y_H3K4 = t(CMC2_H3K4me1[,match(indis_NI,indis_H3K4me1)+2])
Y_ATAC = t(CMC2_ATACseq[,match(indis_NI,indis_ATAC)+2])Increase 200kb window to 2000kb to reduce the size of matrix
Y_RNAr = do.call(cbind, by(t(Y_RNA), (seq(ncol(Y_RNA)) - 1) %/% 10, FUN = colSums))
Y_H3K4r = do.call(cbind, by(t(Y_H3K4), (seq(ncol(Y_H3K4)) - 1) %/% 10, FUN = colSums))
Y_ATACr = do.call(cbind, by(t(Y_ATAC), (seq(ncol(Y_ATAC)) - 1) %/% 10, FUN = colSums))library(flashier)
Y = cbind(Y_RNAr,Y_H3K4r,Y_ATACr)
dim(Y)
Y = log(Y+1e-4)
# fit_flash = flashier::flash(Y,var.type = 1)
# saveRDS(fit_flash,'output/flash_on_cmc2_reduced_2000kb.rds')
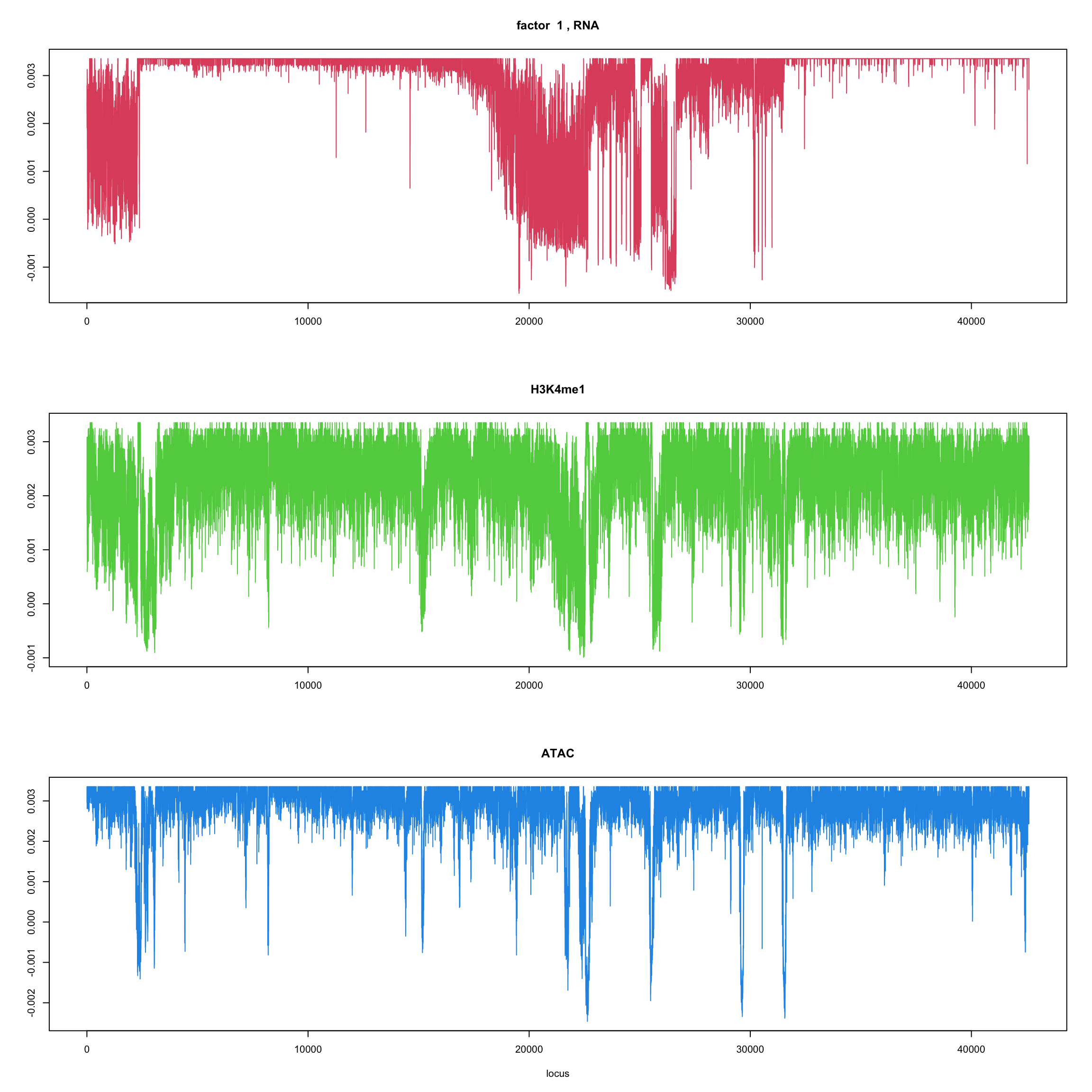
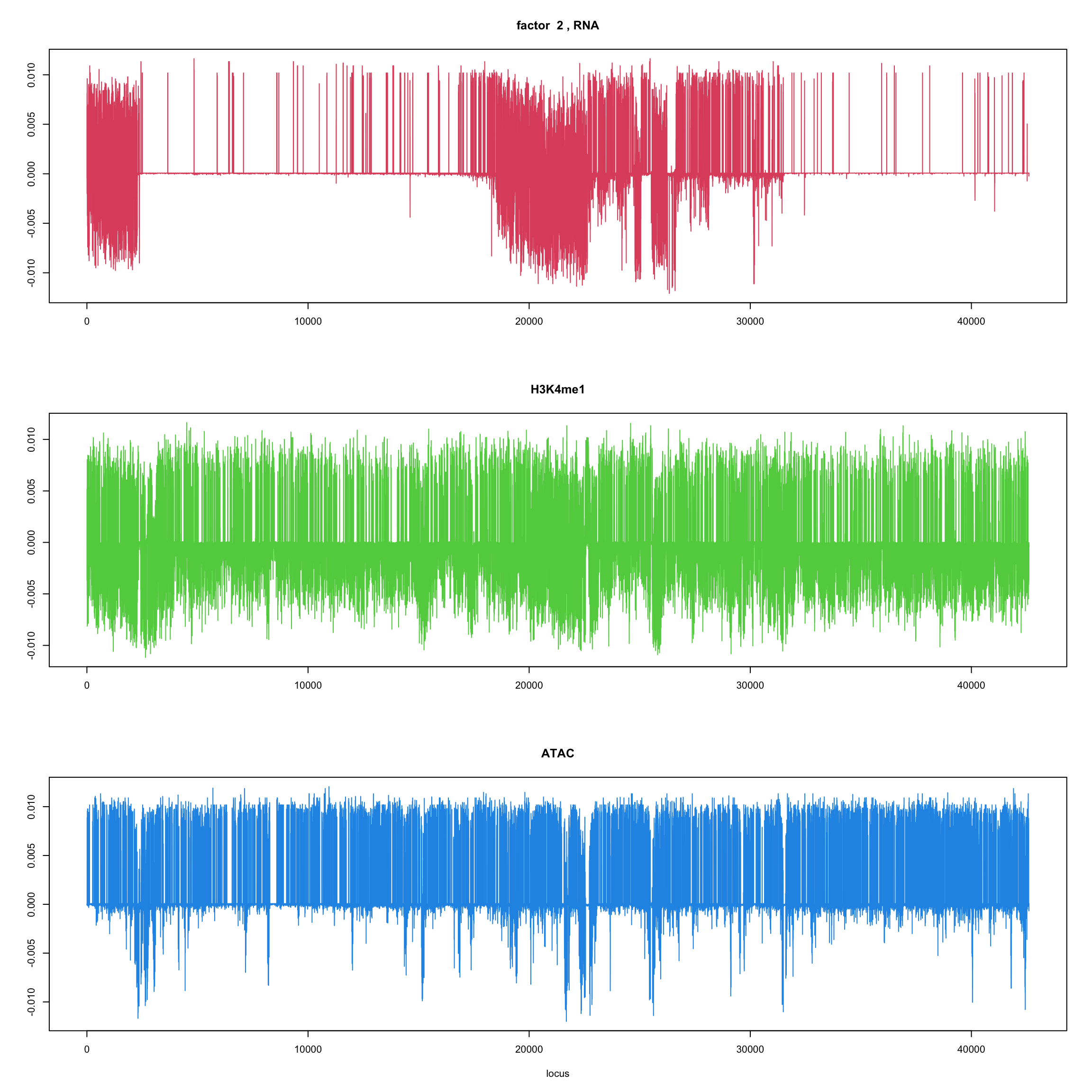
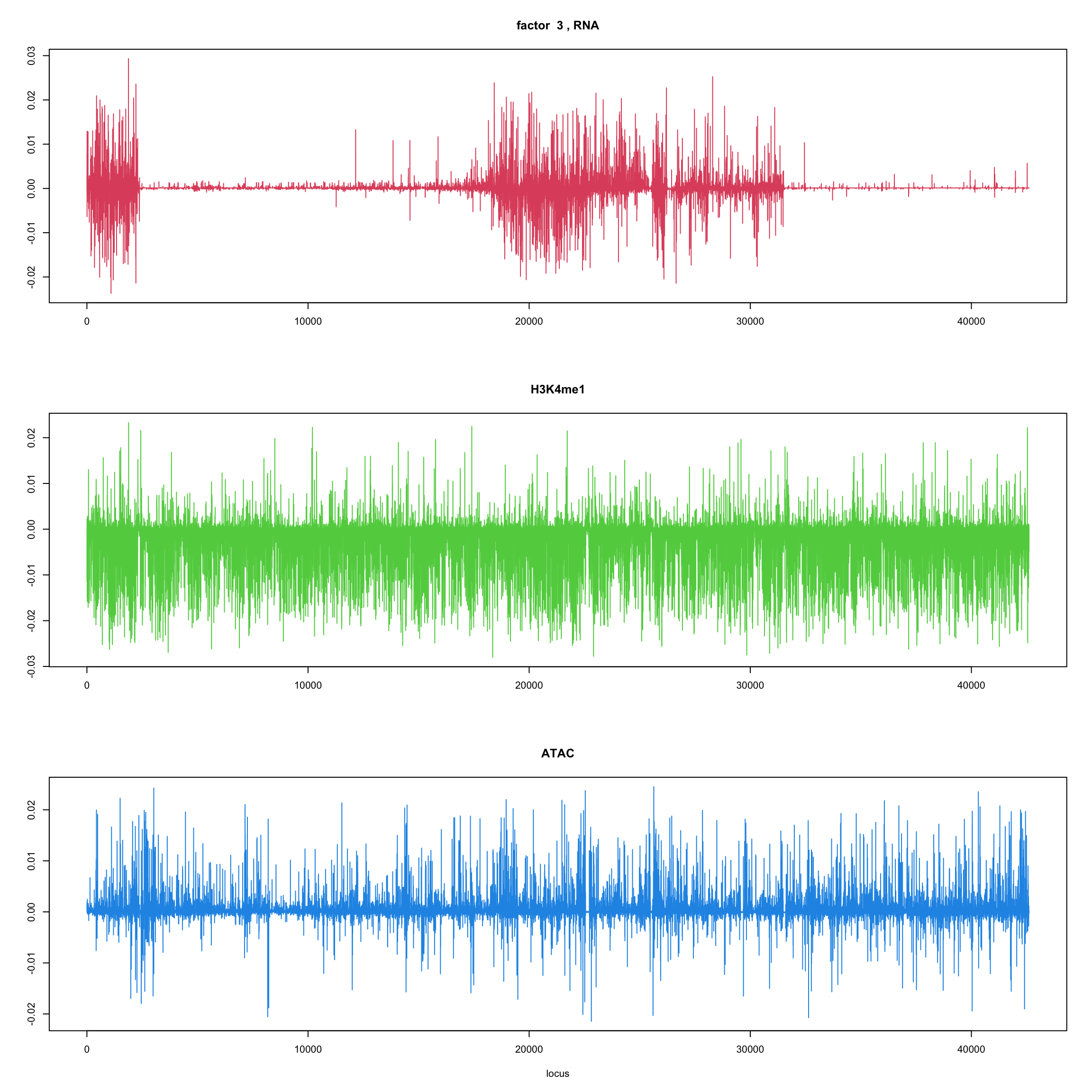
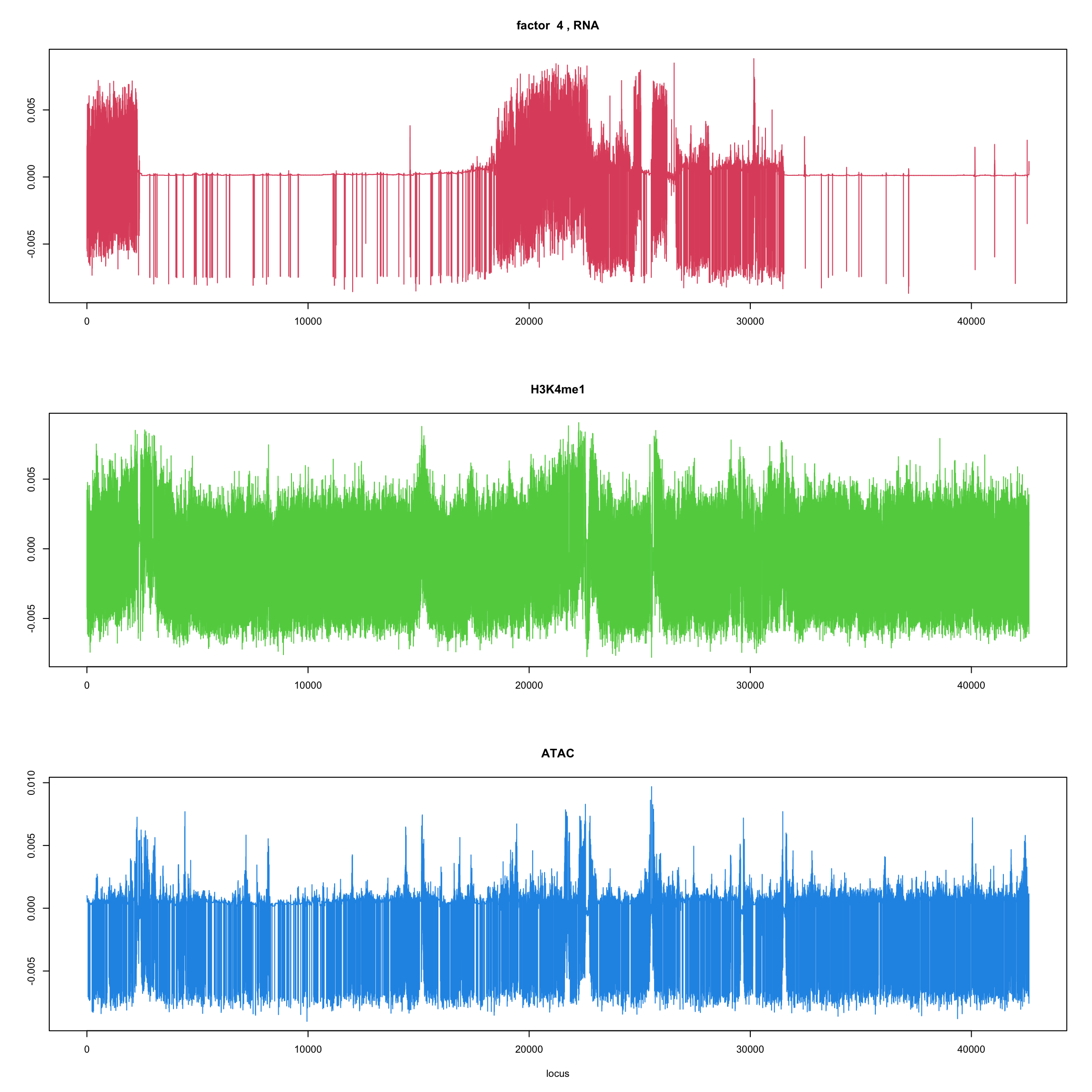
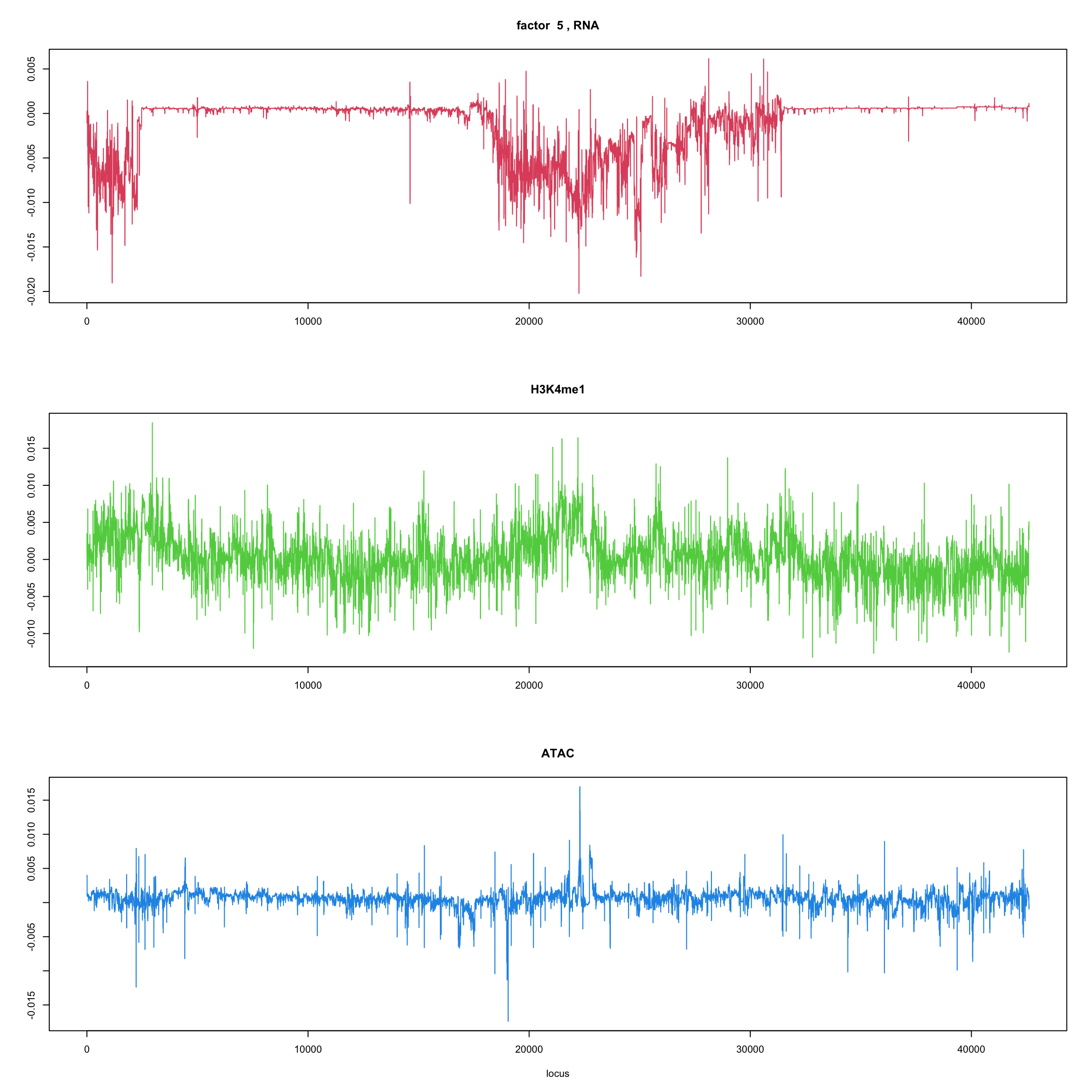
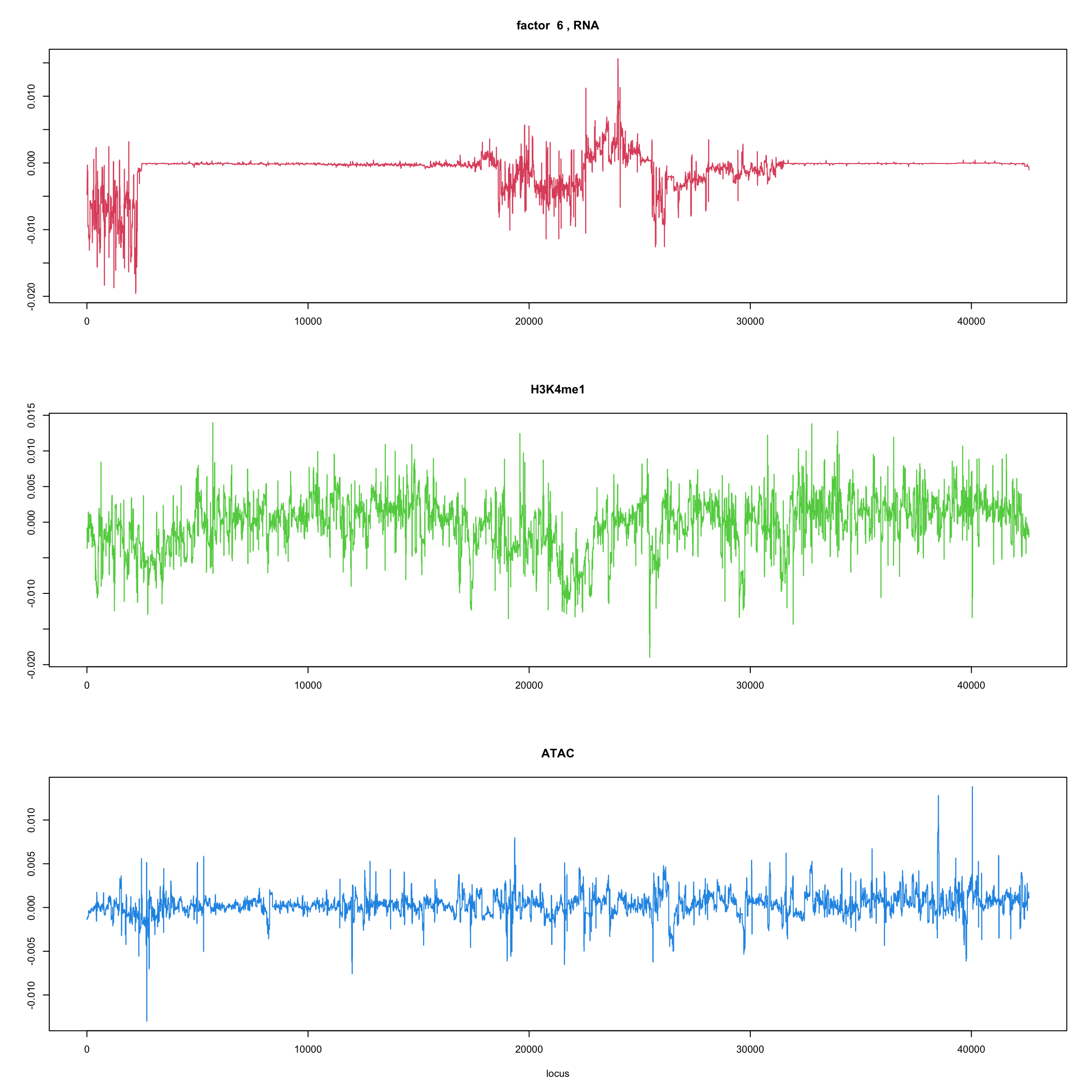
#fit_flash_by_row = flashier::flash(Y,var.type = 1,greedy.Kmax=5)ploter.flash.factor = function(fit_flash,k=1,p){
#plot(fit_flash$loadings.pm[[2]][,k],type='l',xlab='locus',ylab='',main=paste('factor ',k))
par(mfrow=c(3,1))
plot(fit_flash$loadings.pm[[2]][1:(p/3),k],type='l',
xlab='',ylab='',main=paste('factor ',k, ', RNA'),col=2)
plot(fit_flash$loadings.pm[[2]][((p/3+1):(p/3*2)),k],type='l',
xlab='',ylab='',main='H3K4me1',col=3)
plot(fit_flash$loadings.pm[[2]][((p/3*2+1):(p)),k],type='l',
xlab='locus',ylab='',main='ATAC',col=4)
}
ploter.flash.loading = function(fit_flash,K){
par(mfrow=c(ceiling(K/2),2))
for(k in 1:K){
plot(fit_flash$loadings.pm[[1]][,k],type='h',xlab='individuals',ylab='',main=paste('loading ',k))
}
}fit_flash = readRDS('output/flash_on_cmc2_reduced_2000kb.rds')
plot(fit_flash$pve,type='p',pch=20,xlab = 'k',ylab='pve')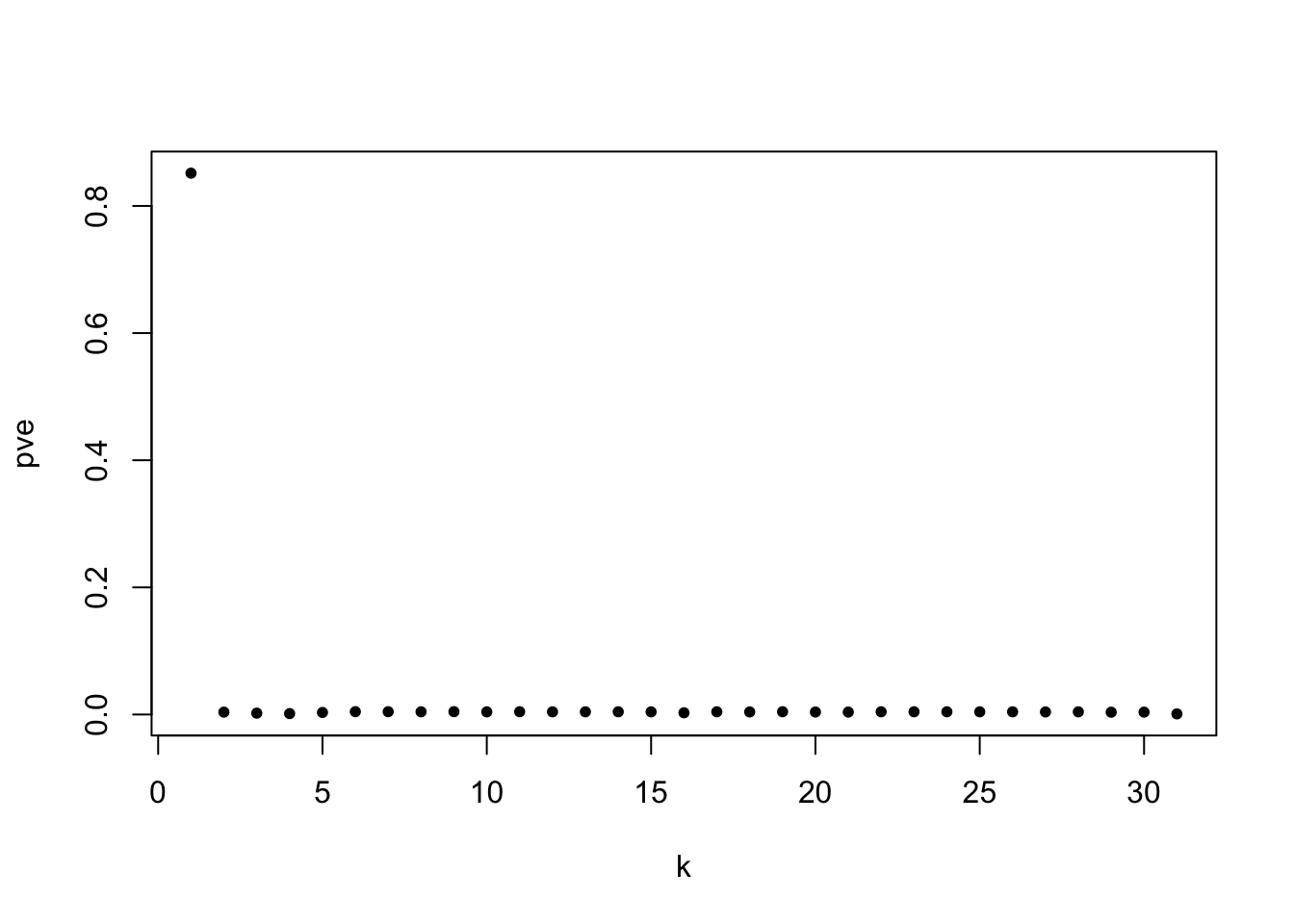
ploter.flash.loading(fit_flash,10)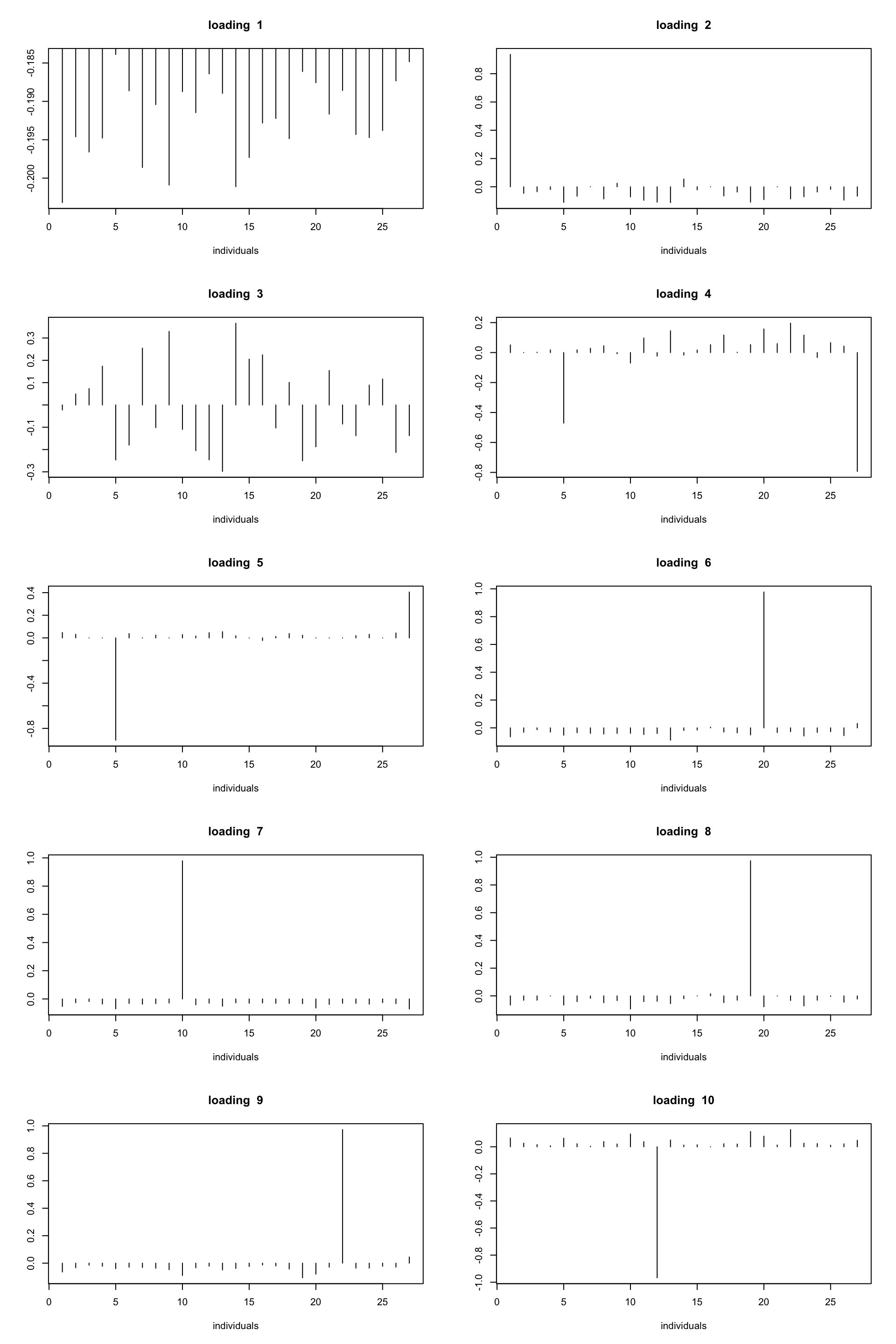
for(k in 1:10){
ploter.flash.factor(fit_flash,k,127827)
}
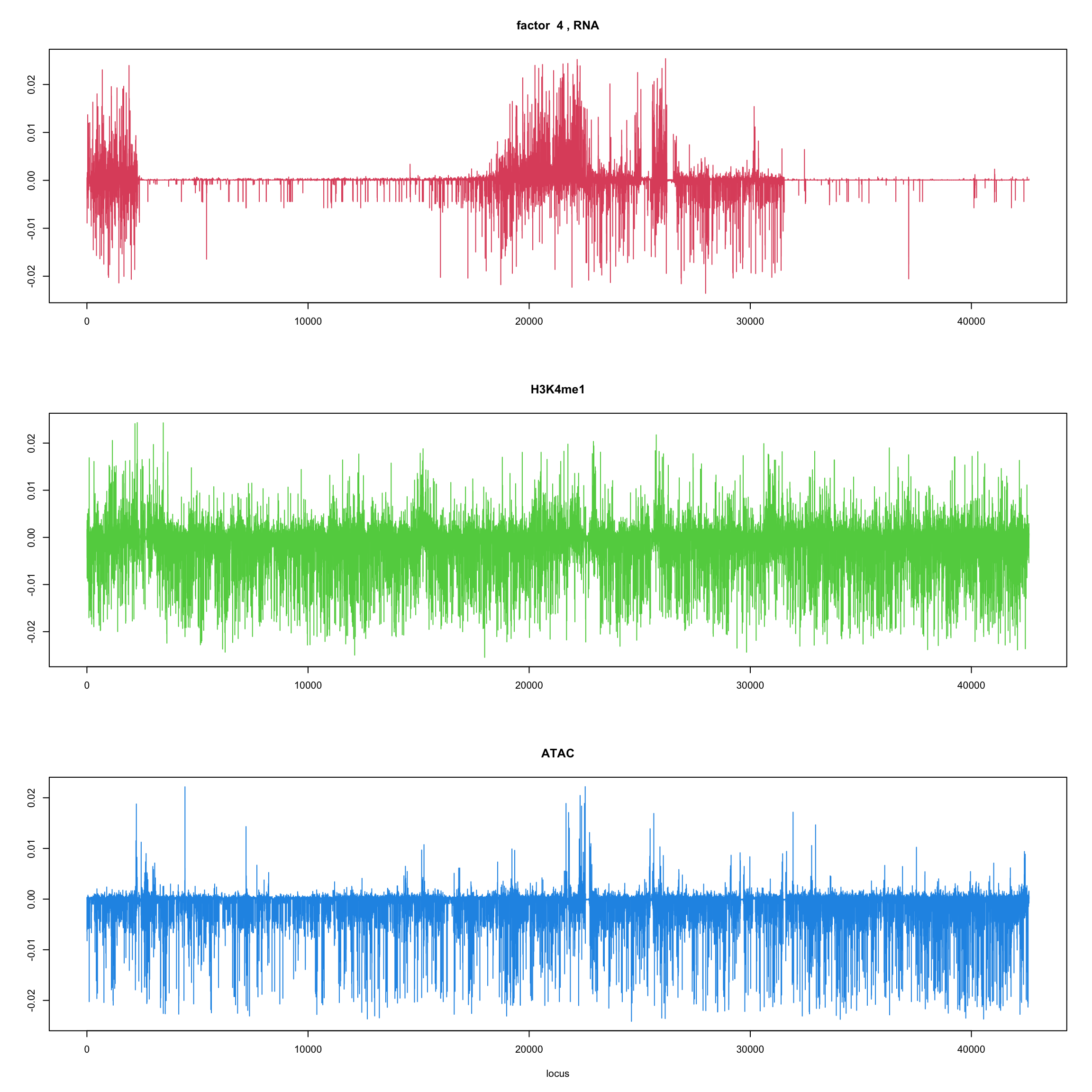
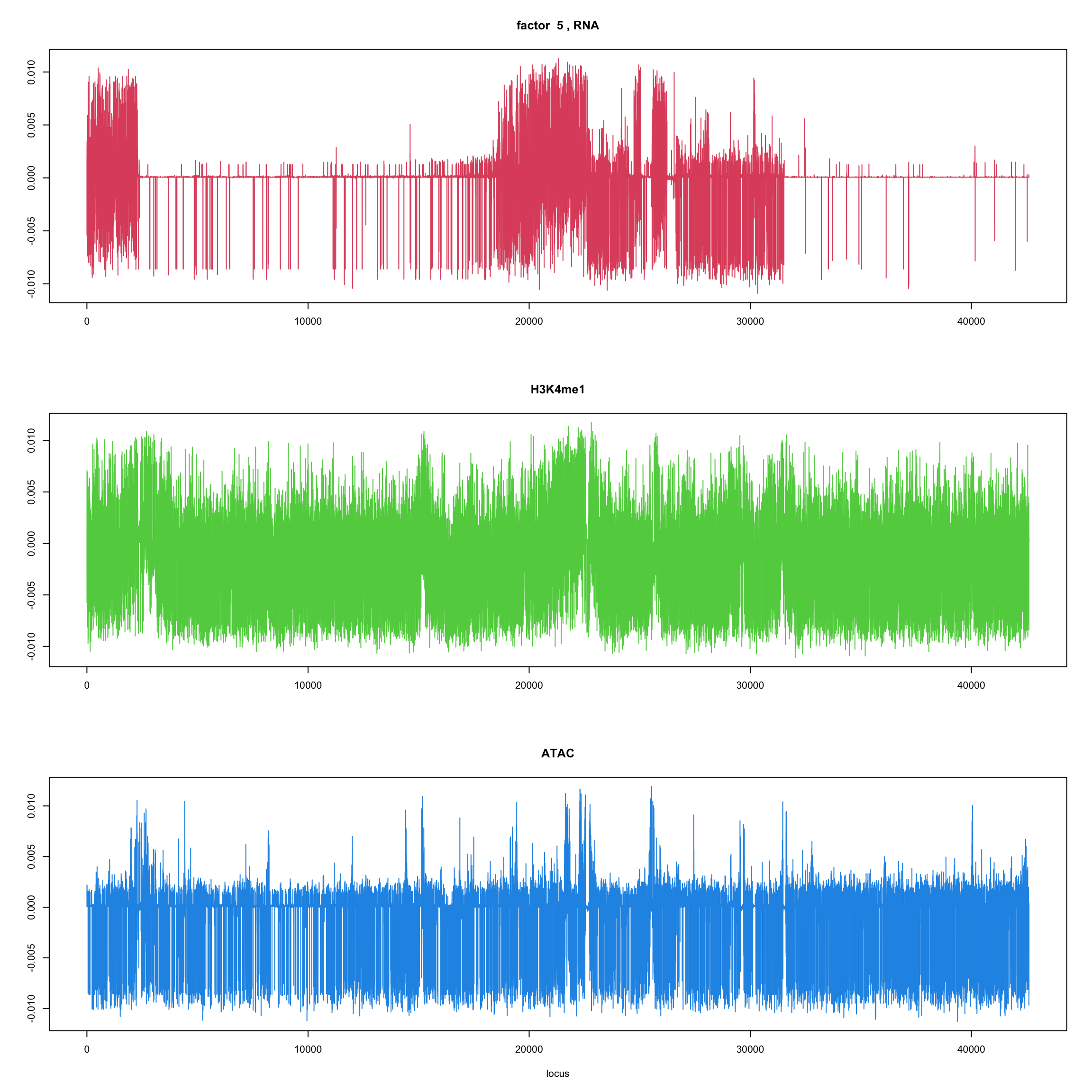
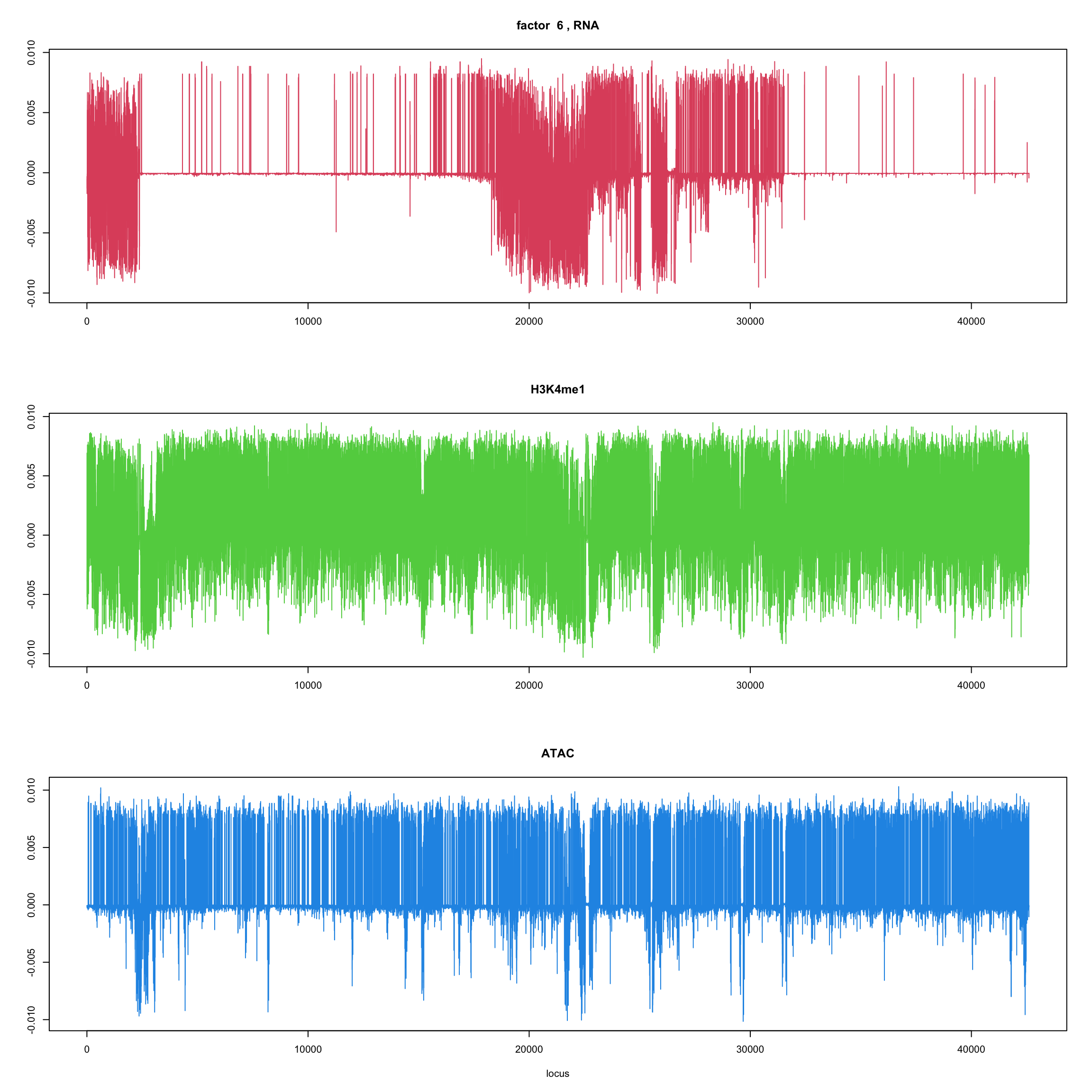
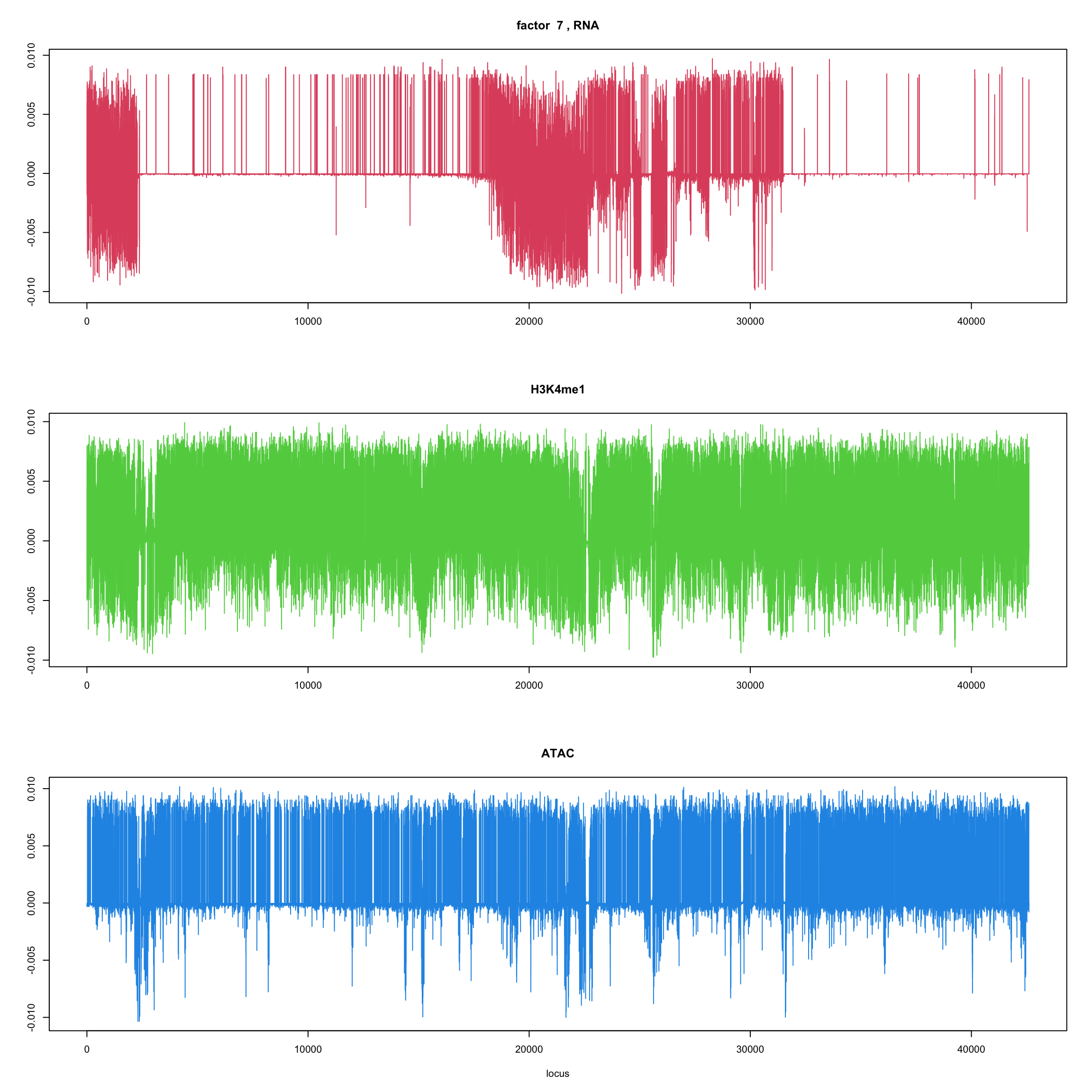
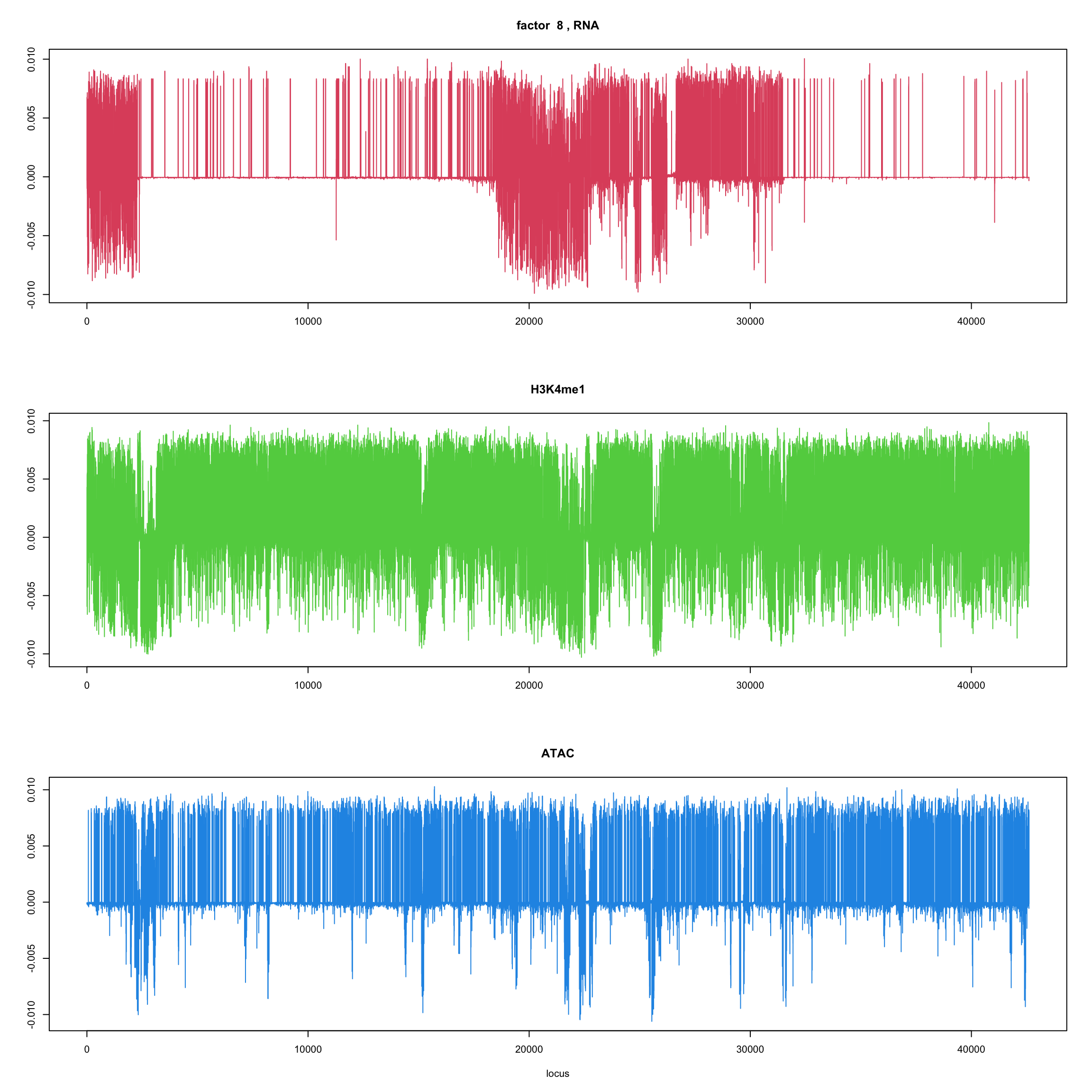
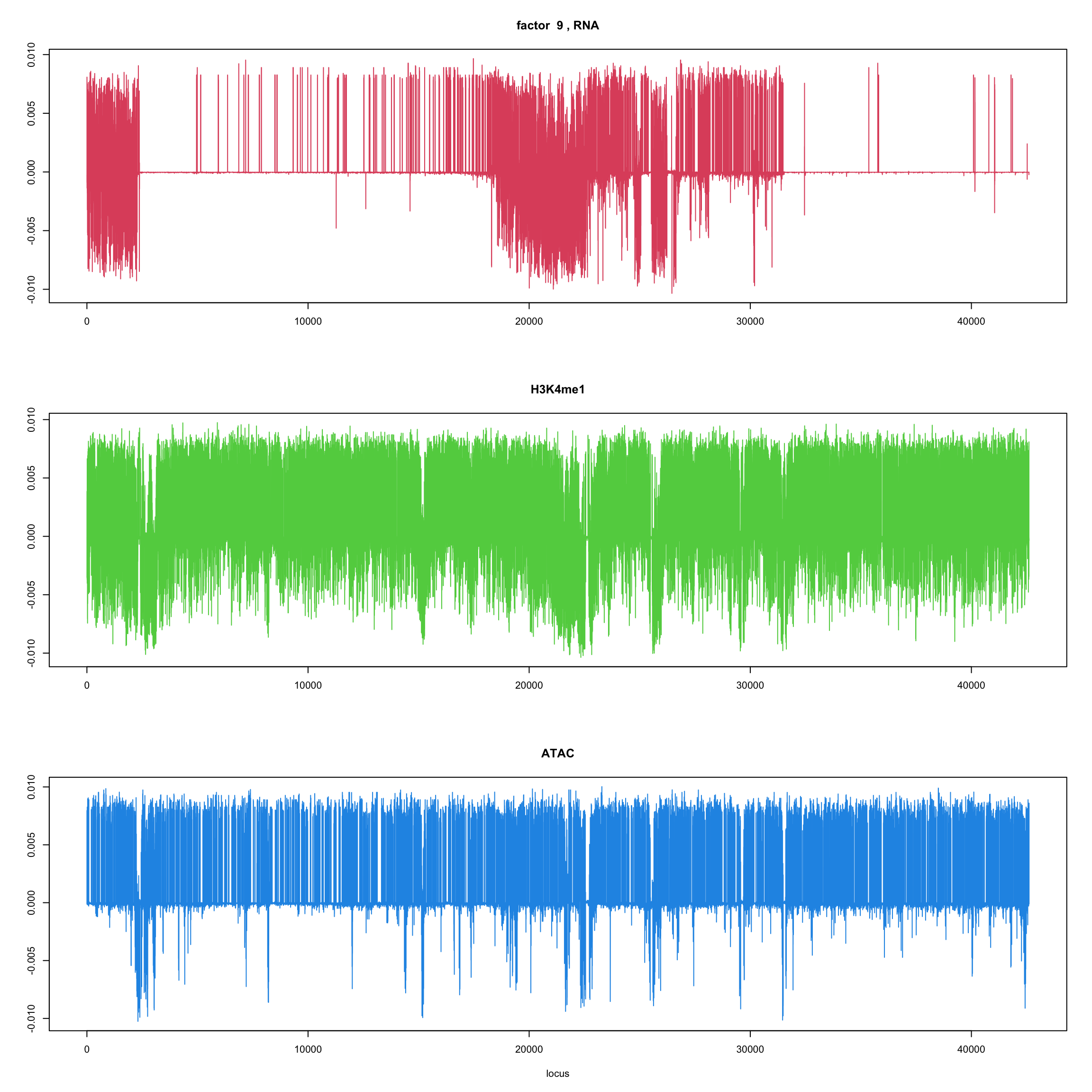
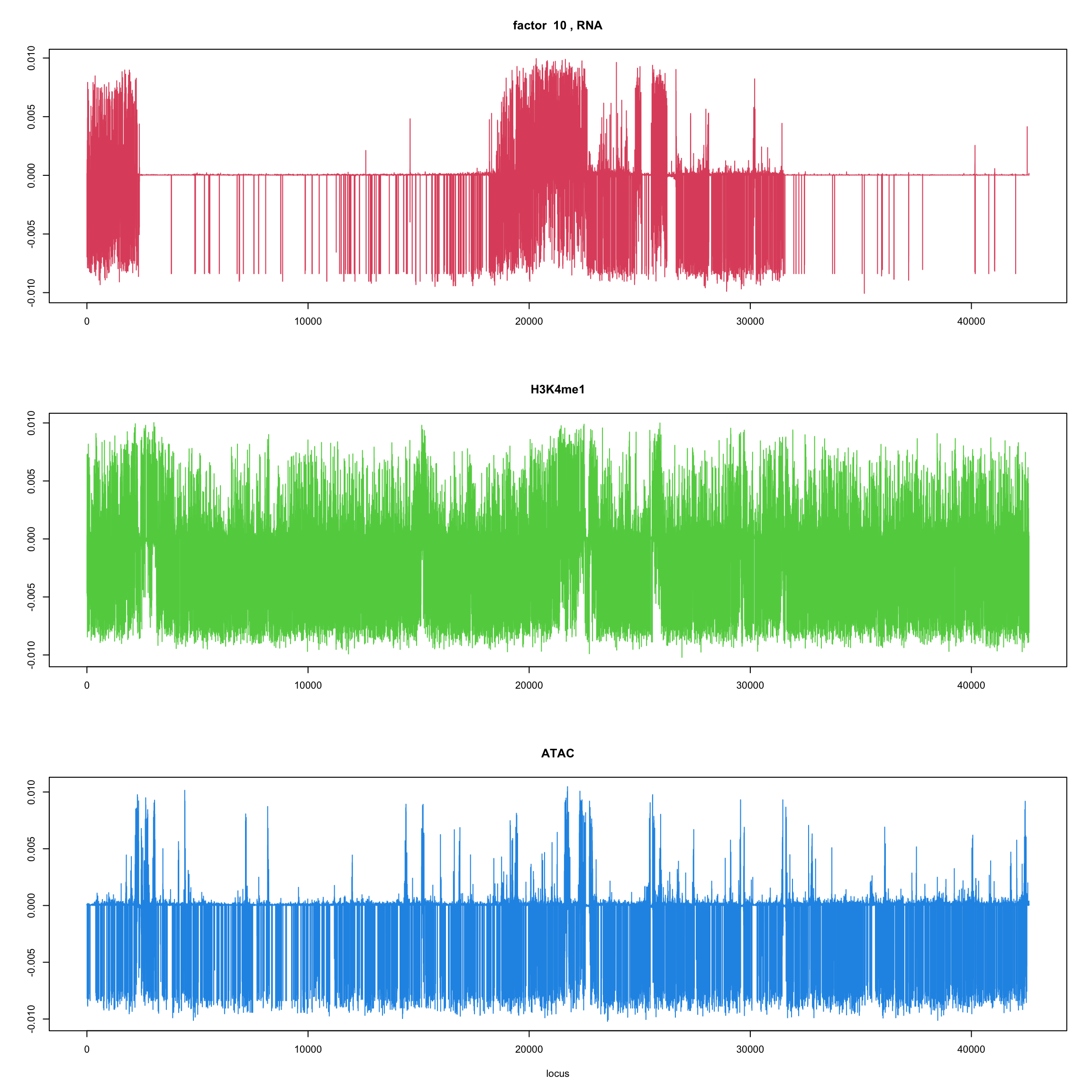
Try to fit the wave ebmf
source('code/wave_ebmf.R')Loading required package: MASSWaveThresh: R wavelet software, release 4.6.8, installedCopyright Guy Nason and others 1993-2016Note: nlevels has been renamed to nlevelsWTLoading flashrRegistered S3 method overwritten by 'flashr':
method from
print.flash flashier#rm.idx = c(1:4921,(42609-4920+1):42609)
#Y = cbind(Y_RNAr[,-rm.idx],Y_H3K4r[,-rm.idx],Y_ATACr[,-rm.idx])
# fit_wave = wave_ebmf(Y,Kmax=10,s=fit_flash$residuals.sd)
# saveRDS(fit_wave,'output/wave_flash_on_cmc2_reduced_2000kb.rds')
fit_wave = readRDS('output/wave_flash_on_cmc2_reduced_2000kb.rds')
par(mfrow=c(1,1))
plot(fit_wave$pve,type='p',pch=20,xlab = 'k',ylab='pve')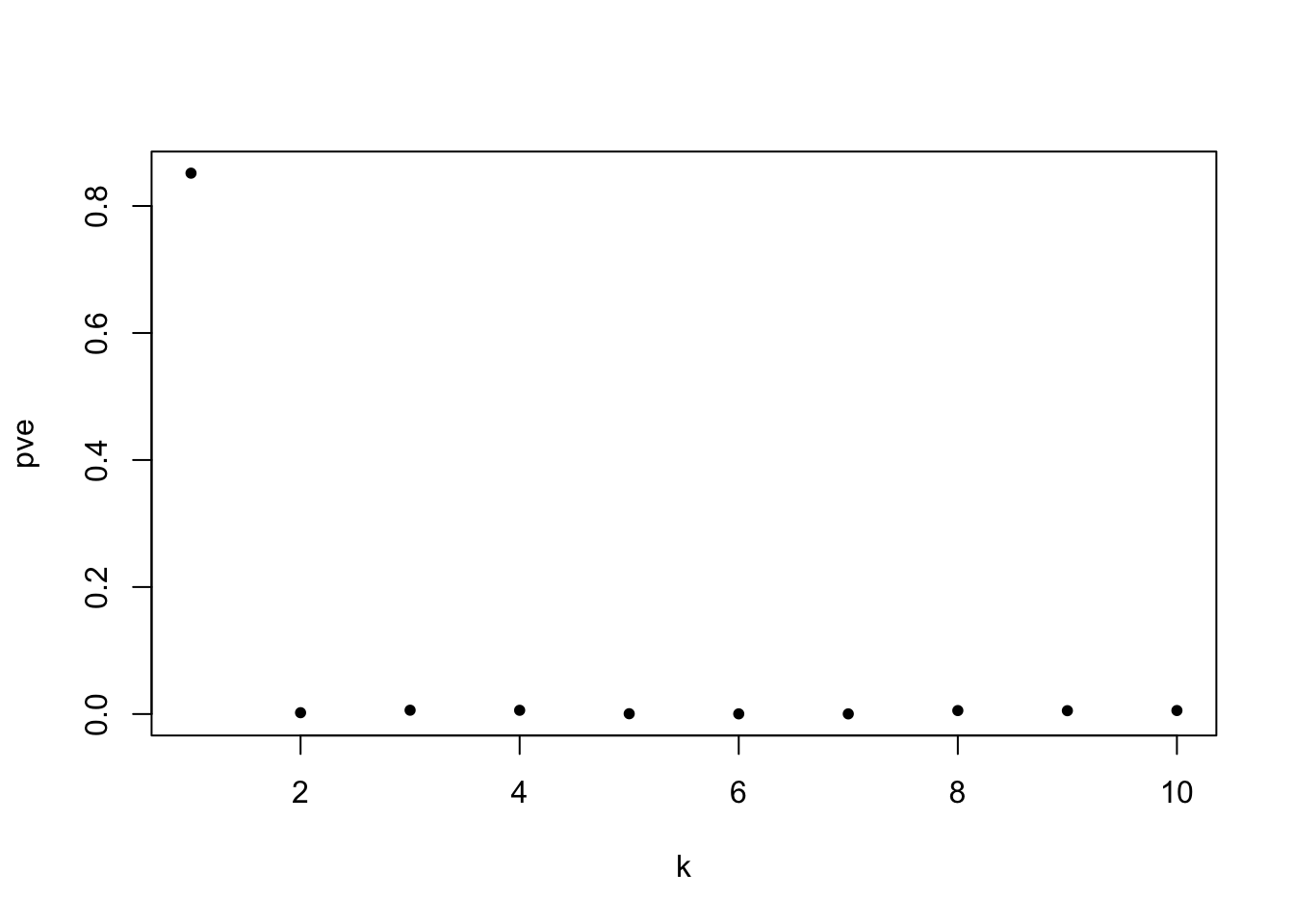
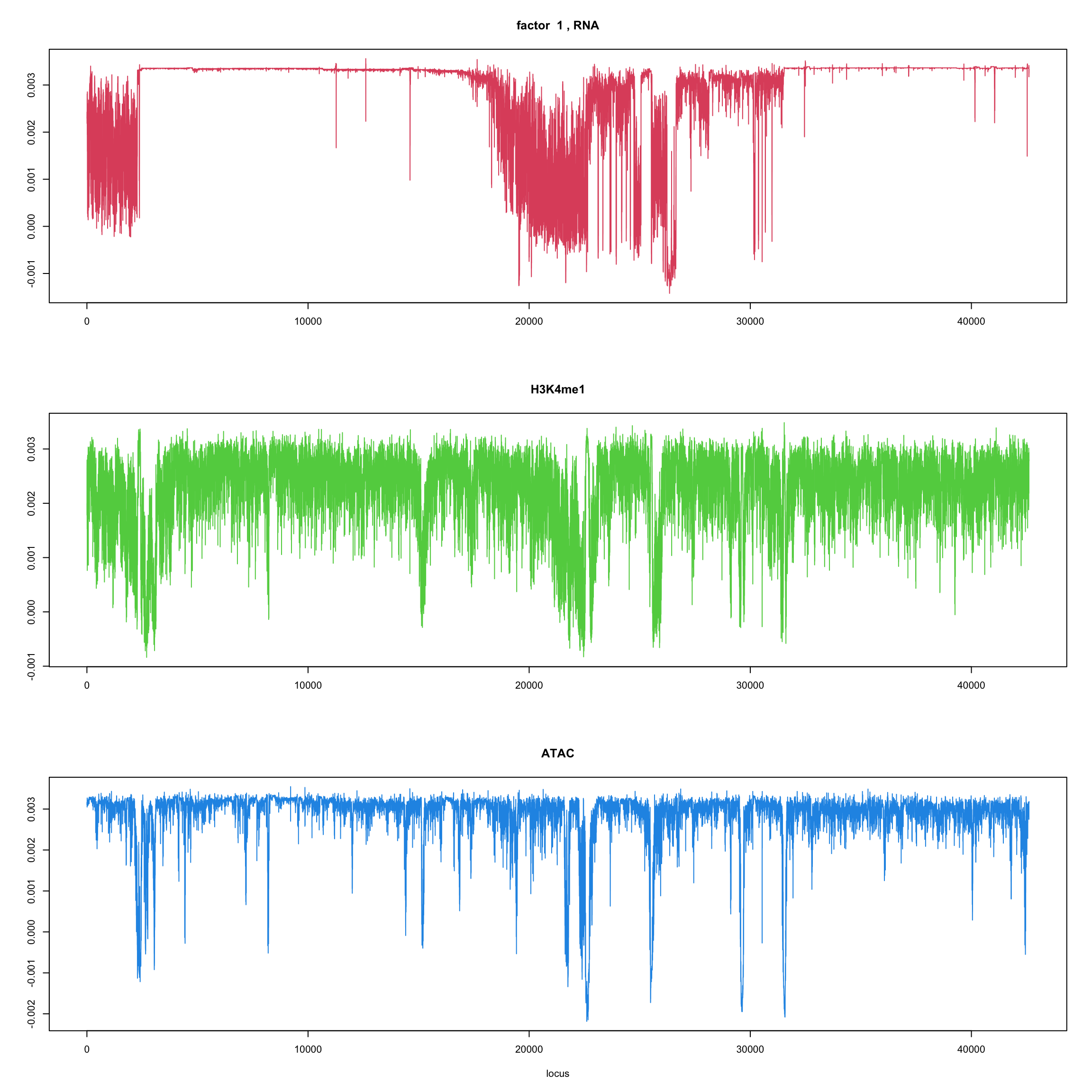
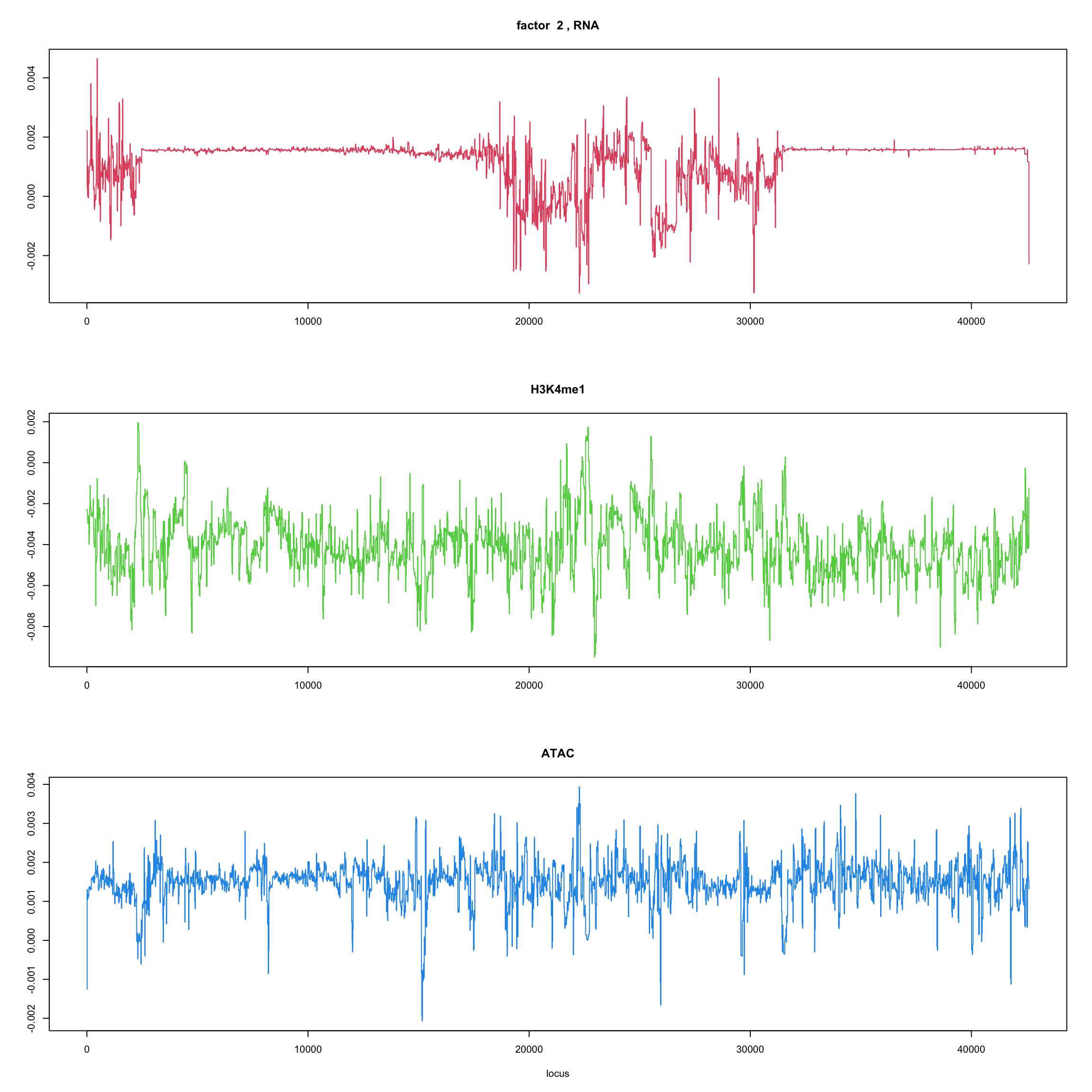
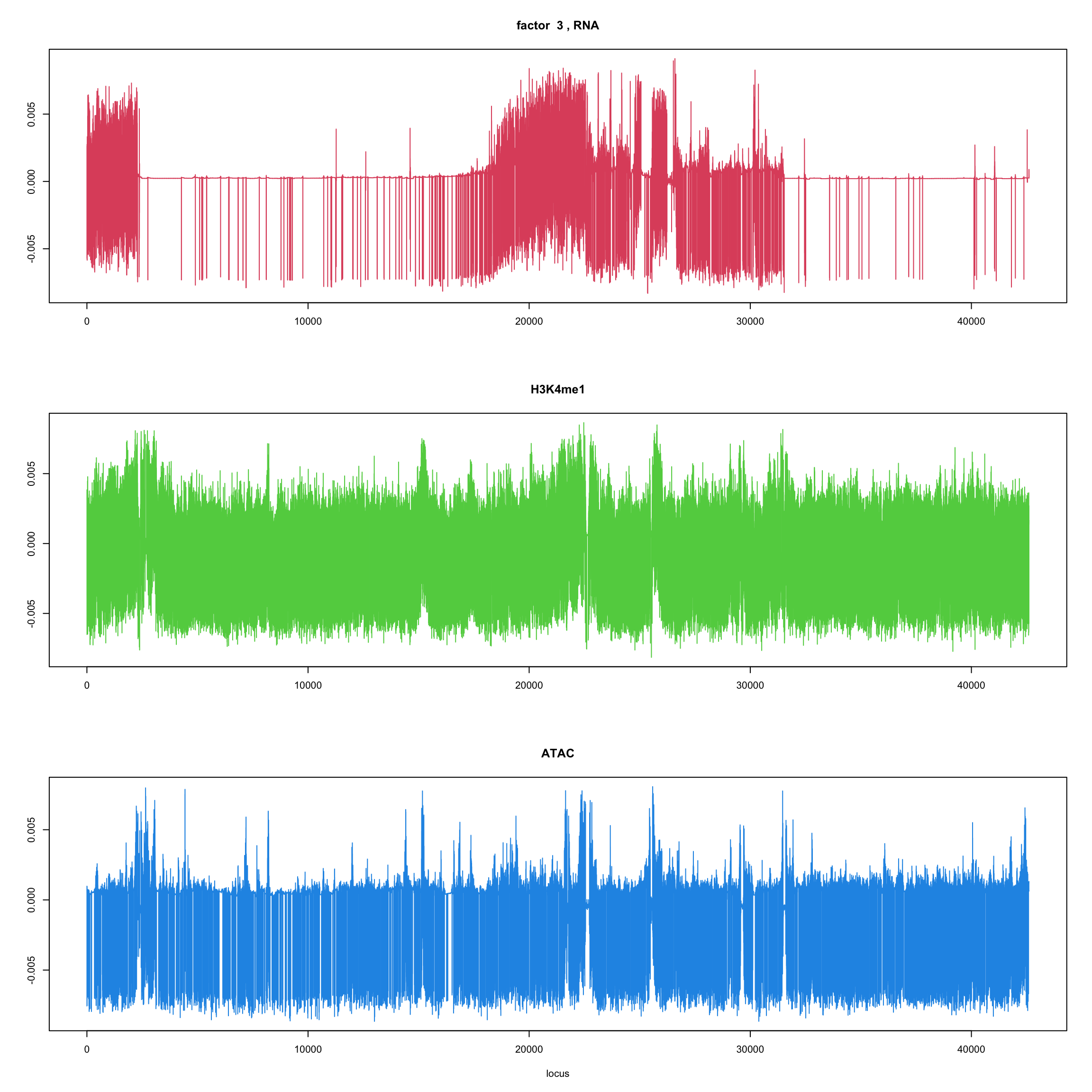
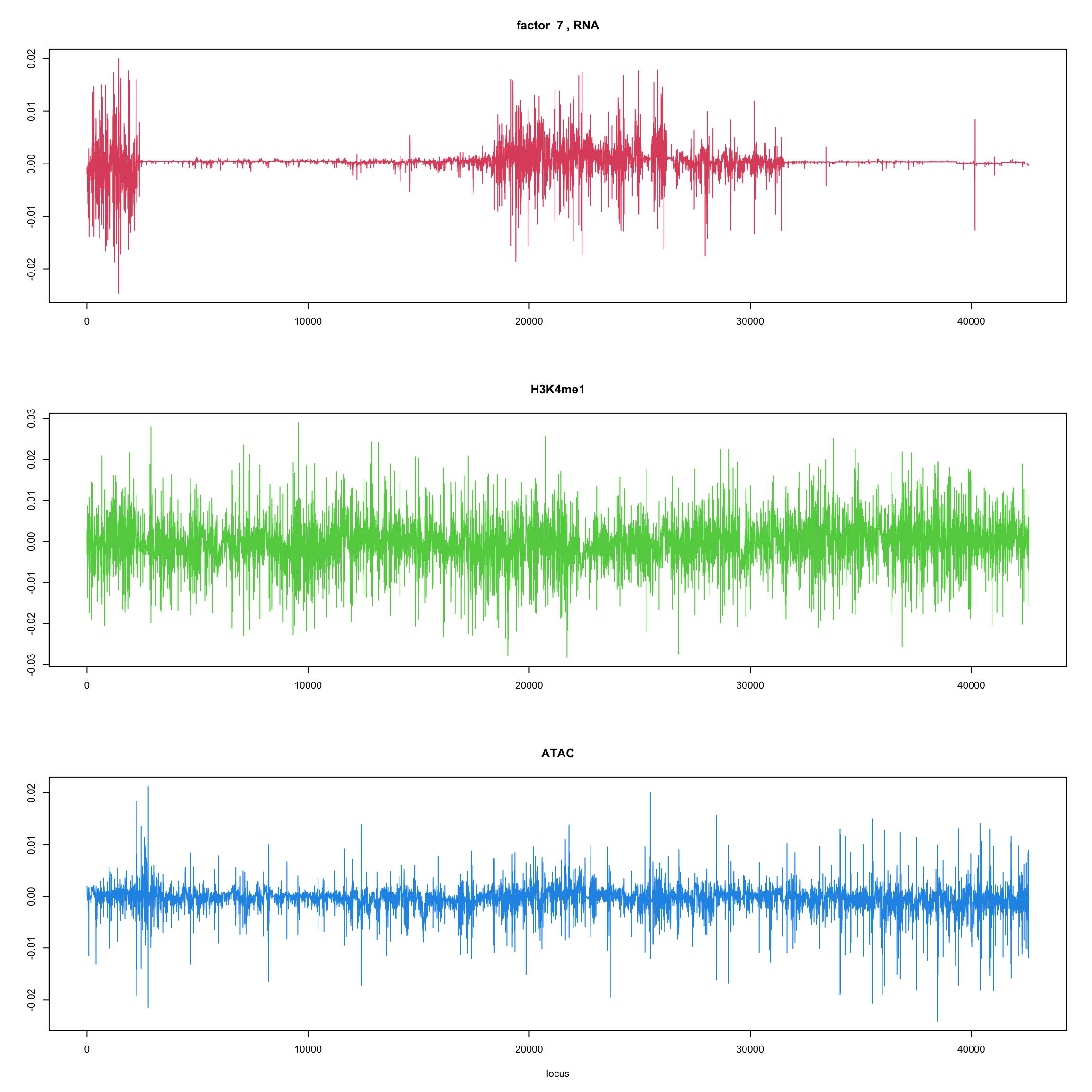
ploter.wave.flash.factor = function(fit_wave,k=1,p){
#plot(fit_flash$loadings.pm[[2]][,k],type='l',xlab='locus',ylab='',main=paste('factor ',k))
par(mfrow=c(3,1))
plot(fit_wave$ldf$f[1:(p/3),k],type='l',
xlab='',ylab='',main=paste('factor ',k, ', RNA'),col=2)
plot(fit_wave$ldf$f[((p/3+1):(p/3*2)),k],type='l',
xlab='',ylab='',main='H3K4me1',col=3)
plot(fit_wave$ldf$f[((p/3*2+1):(p)),k],type='l',
xlab='locus',ylab='',main='ATAC',col=4)
}
ploter.wave.flash.loading = function(fit_wave,K){
par(mfrow=c(ceiling(K/2),2))
for(k in 1:K){
plot(fit_wave$ldf$l[,k],type='h',xlab='individuals',ylab='',main=paste('loading ',k))
}
}ploter.wave.flash.loading(fit_wave,10)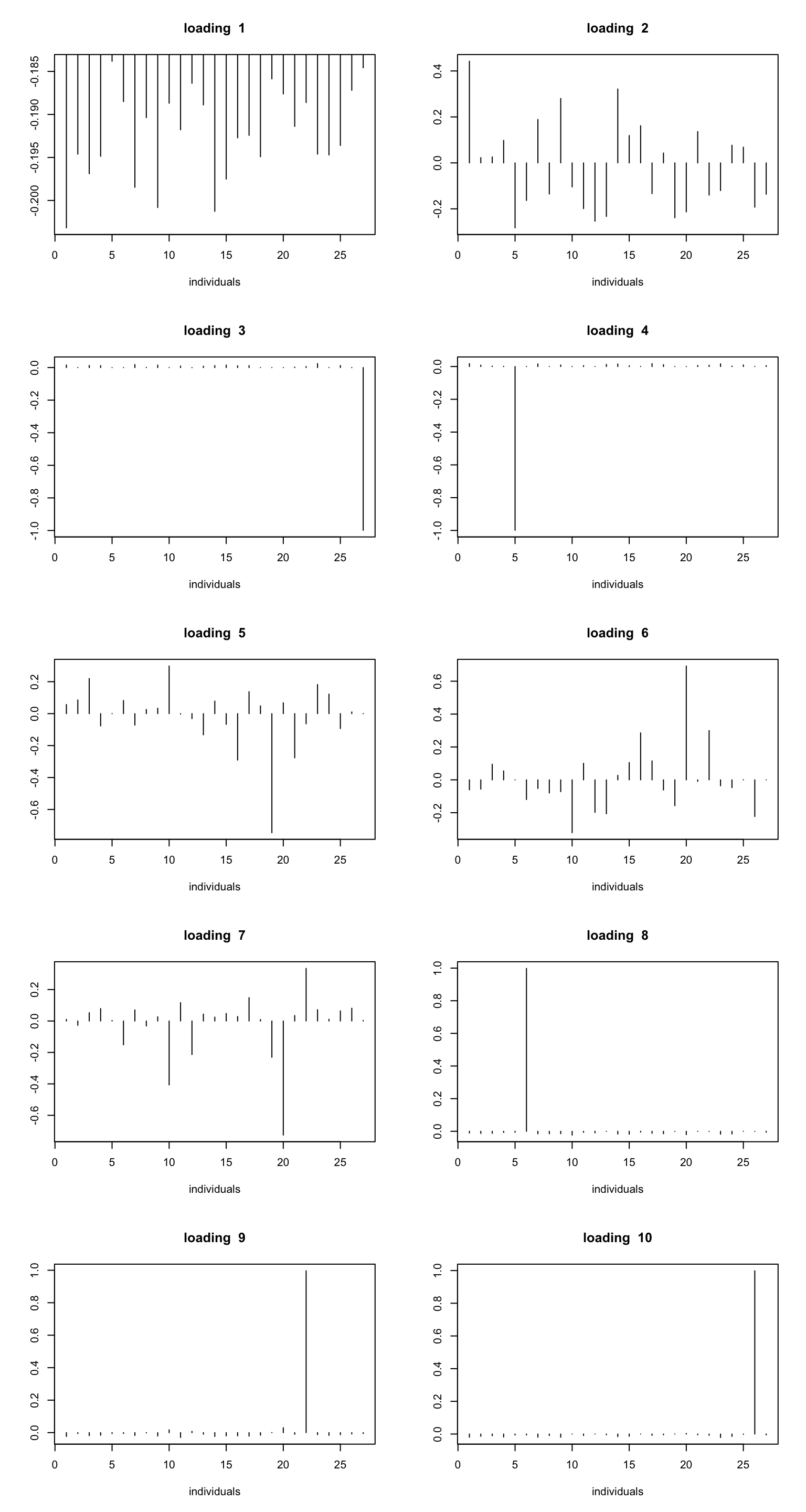
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
for(k in 1:10){
ploter.wave.flash.factor(fit_wave,k,127827)
}
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
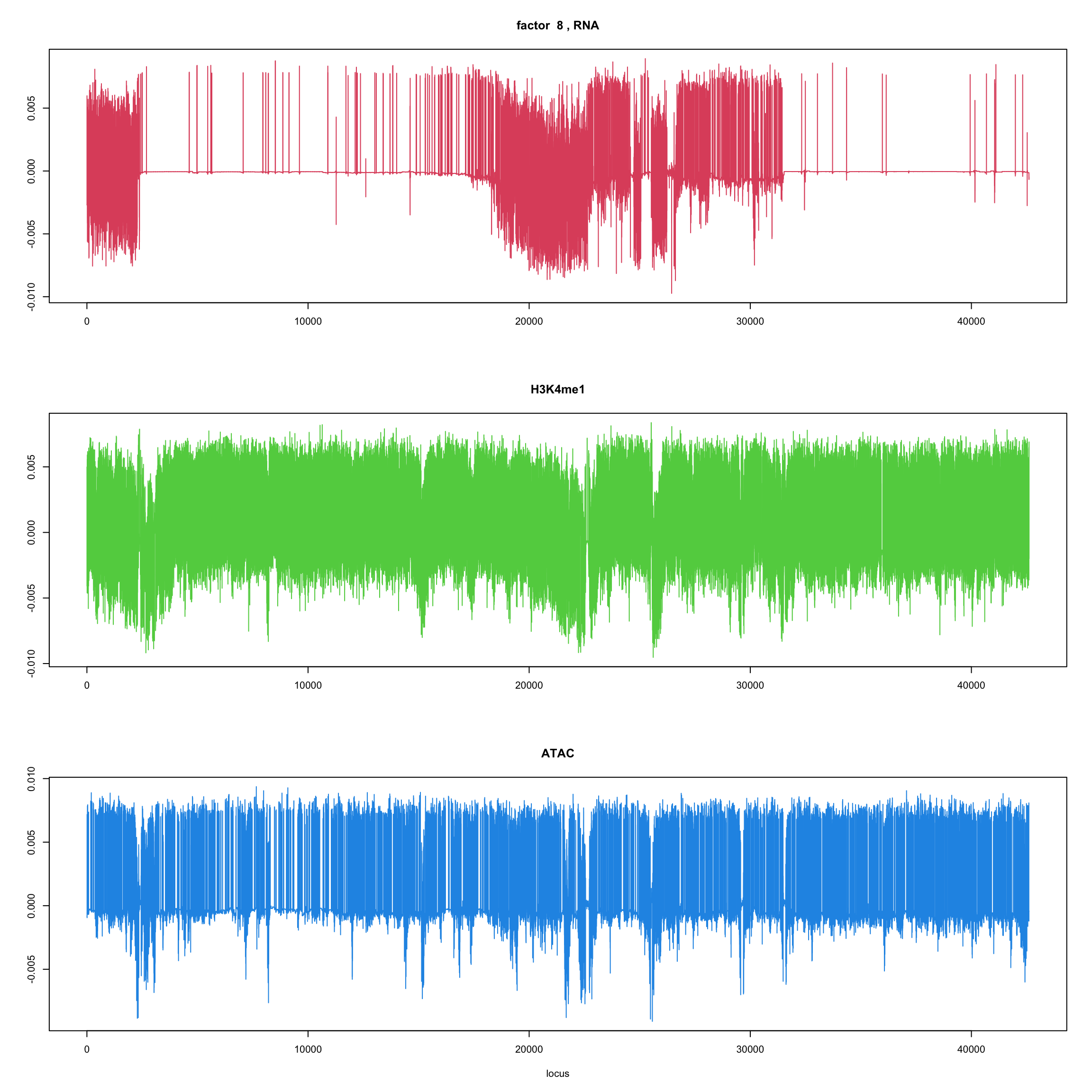
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
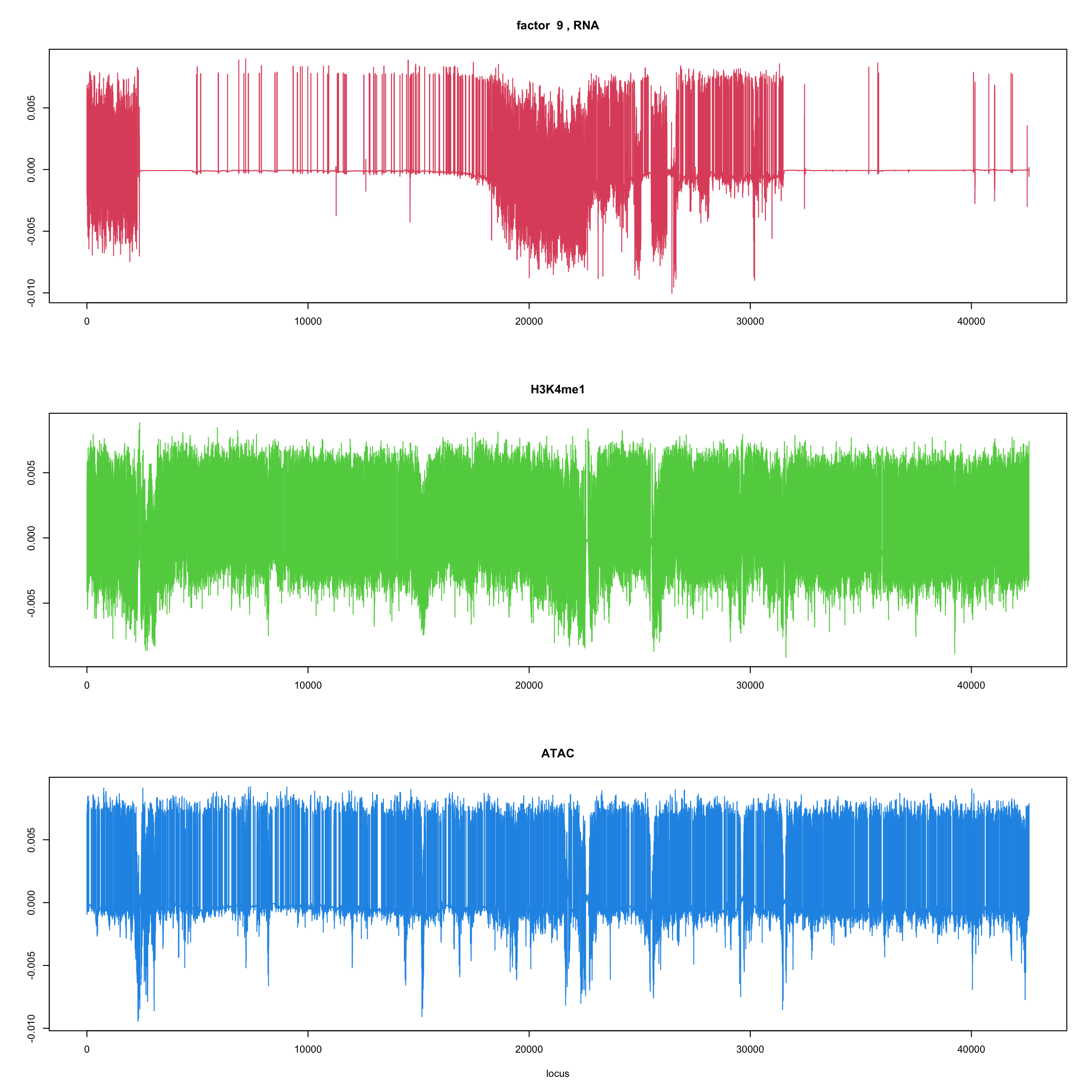
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
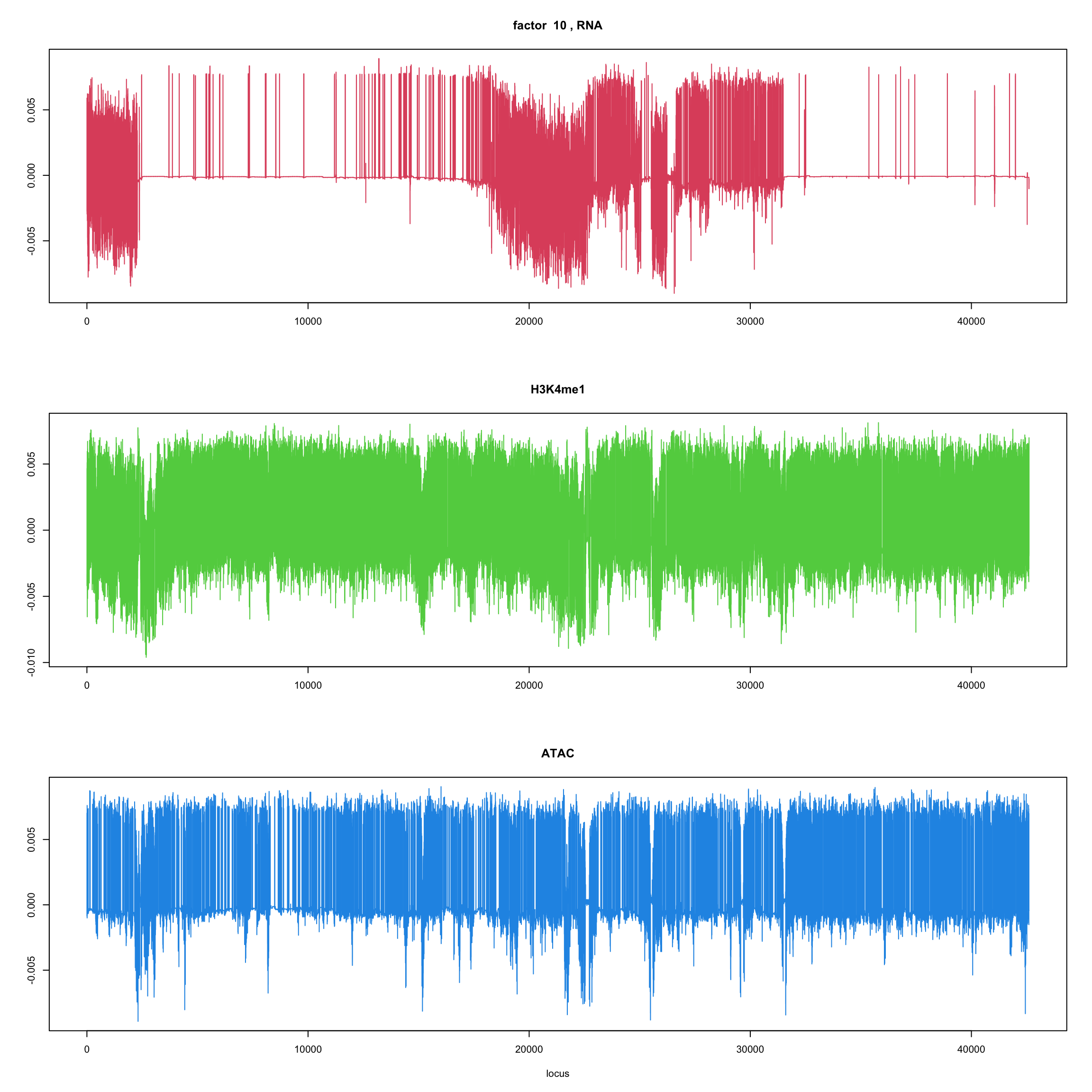
| Version | Author | Date |
|---|---|---|
| ed30472 | Dongyue Xie | 2021-09-10 |
FAM220A
FAM220A is a protein coding gene with 2 exons.
FAM220A_ATACseq = read_csv("data/luis/FAM220A_ATACseq.csv")
FAM220A_RNAseq = read_csv("data/luis/FAM220A_RNAseq.csv")
FAM220A_H3K4me1 = read_csv("data/luis/FAM220A_H3K4me1.csv")
FAM220A_ATACseq = do.call(cbind.data.frame, FAM220A_ATACseq)
FAM220A_RNAseq = do.call(cbind.data.frame, FAM220A_RNAseq)
FAM220A_H3K4me1 = do.call(cbind.data.frame, FAM220A_H3K4me1)
dim(FAM220A_ATACseq)
dim(FAM220A_RNAseq)
dim(FAM220A_H3K4me1)
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] flashr_0.6-7 testthat_3.0.0 wavethresh_4.6.8 MASS_7.3-53
[5] readr_1.4.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] pkgload_1.1.0 splines_4.0.3 assertthat_0.2.1 horseshoe_0.2.0
[5] mixsqp_0.3-43 deconvolveR_1.2-1 yaml_2.2.1 remotes_2.2.0
[9] sessioninfo_1.1.1 ebnm_0.1-50 pillar_1.4.6 backports_1.1.10
[13] lattice_0.20-41 glue_1.4.2 digest_0.6.27 promises_1.1.1
[17] colorspace_1.4-1 plyr_1.8.6 htmltools_0.5.1.1 httpuv_1.5.4
[21] Matrix_1.2-18 pkgconfig_2.0.3 devtools_2.3.2 invgamma_1.1
[25] purrr_0.3.4 scales_1.1.1 processx_3.5.1 whisker_0.4
[29] later_1.1.0.1 git2r_0.27.1 tibble_3.0.4 generics_0.1.0
[33] ggplot2_3.3.2 usethis_1.6.3 ellipsis_0.3.1 withr_2.3.0
[37] ashr_2.2-47 cli_2.4.0 magrittr_2.0.1 crayon_1.3.4
[41] memoise_1.1.0 evaluate_0.14 ps_1.4.0 fs_1.5.0
[45] truncnorm_1.0-8 pkgbuild_1.1.0 tools_4.0.3 prettyunits_1.1.1
[49] softImpute_1.4 hms_0.5.3 REBayes_2.2 lifecycle_1.0.0
[53] stringr_1.4.0 munsell_0.5.0 trust_0.1-8 flashier_0.2.9
[57] irlba_2.3.3 callr_3.6.0 compiler_4.0.3 rlang_0.4.10
[61] grid_4.0.3 rstudioapi_0.11 rmarkdown_2.5 gtable_0.3.0
[65] DBI_1.1.0 reshape2_1.4.4 R6_2.4.1 knitr_1.30
[69] dplyr_1.0.5 rprojroot_1.3-2 desc_1.2.0 stringi_1.5.3
[73] parallel_4.0.3 SQUAREM_2020.5 Rcpp_1.0.5 vctrs_0.3.7
[77] tidyselect_1.1.0 xfun_0.18